





CONCEPTO Y RESEÑA HISTORICA
La micosis fungoide (MF) es un linfoma cutáneo de células T epidermotropo, caracterizado por una proliferación de linfocitos T neoplásicos de pequeño o mediano tamaño con núcleo cerebriforme (1). Se manifiesta inicialmente en la piel y suele permanecer en ella durante años o décadas, para posteriormente, en fases avanzadas, afectar a ganglios linfáticos y órganos internos, pudiendo ocasionar la muerte del paciente (1-3).
El síndrome de Sézary (SS) es una variante de linfoma cutáneo de células T y está definido históricamente por la tríada de eritrodermia, linfadenopatías y presencia de células neoplásicas (células de Sézary) en piel, ganglios linfáticos y sangre periférica (1, 4).
La MF fue descrita inicialmente por Alibert en 1806 (5), denominándola de esta manera porque las lesiones adoptaban un aspecto semejante a hongos. Posteriormente, Bazin en 1870 (6) dio individualidad propia a la enfermedad e hizo su descripción clínica con los tres estadios evolutivos clásicos (premicósico, infiltrativo y tumoral). En 1938, Sézary y Bouvrain (4) describieron la tríada clínica de eritrodermia, linfadenopatías y células atípicas circulantes en sangre periférica, lo que hoy conocemos como síndrome de Sézary.
CLASIFICACIONES
Hasta fechas próximas los linfomas cutáneos fueron clasificados de acuerdo a las clasificaciones histológicas realizadas por hematólogos y patólogos para los linfomas no Hodgkin ganglionares. En este sentido se utilizaron durante muchos años la clasificación de Kiel modificada (7) y la Working-Formulation (8). La aparición en los últimos años de nuevos tipos de linfomas y la necesidad de utilizar de manera uniforme una misma clasificación por los distintos grupos de trabajo dio lugar a que el Grupo Internacional de Estudio de Linfoma propusiera en 1994 una nueva clasificación denominada «Clasificación Revisada Europea-Americana de Neoplasias Linfoides» (REAL) (9), modificada posteriormente por la OMS en 1999 (10).
Recientemente, y apoyándose en la idea de que los linfomas cutáneos primarios deben de ser considerados un grupo distintivo clínica y biológicamente, se publicó la clasificación de linfomas cutáneos primarios por el Grupo de Estudio del Linfoma Cutáneo de la Organización Europea para la Investigación y Tratamiento del Cáncer (EORTC) (1) (tabla I). Esta clasificación se basó en una combinación de criterios clínicos, histológicos, inmunohistoquímicos y genéticos, diferenciando las distintas entidades sobre la base de unos criterios que en conjunto nos permitirían predecir el curso clínico, la respuesta al tratamiento y el pronóstico.

EPIDEMIOLOGÍA
La MF representa el tipo más frecuente de linfoma cutáneo primario (1, 3). Se considera que existe un aumento progresivo en su incidencia (11), aunque no sabemos si este incremento es debido a un aumento real o, por el contrario, sólo refleja un mejor conocimiento de la entidad y un diagnóstico más temprano (3). Weinstock y Horm (11) hallaron una incidencia media de 0,3 casos por 100.000 habitantes, representando la MF el 2,2% de todos los linfomas.
La incidencia de la MF se incrementa conforme avanza la edad y la edad media de presentación es aproximadamente a los 50 años (2). Existen publicaciones en niños y adolescentes, sin que por ello comporte peor pronóstico (12-16). Aparece con mayor frecuencia en varones que en mujeres en una relación de 2,2 varones por una mujer, y la raza negra está más frecuentemente afectada que la blanca en una proporción de 2 a 1 (17).
ETIOLOGÍA
La etiología de la MF no se conoce. Se han barajado diversas teorías, entre las que destacamos las siguientes:
— Virus. Aunque han sido varios los virus implicados en la etiología, como el virus del herpes simple o el de Epstein-Barr (3, 18), los datos más relevantes relacionaron a la MF con los retrovirus y, en concreto, con el virus linfotropo de células T humanas (HTLV 1 y 2) (19, 20). En la actualidad existe cierta controversia, ya que algunos estudios han descartado que el virus HTLV 1 pudiera estar implicado directamente en la etiología o patogénesis del linfoma cutáneo de células T (21, 22).
— Estimulación antigénica crónica y persistente. Se ha sugerido que la presencia y persistencia de un antígeno, hasta ahora desconocido, causaría una estimulación persistente de los linfocitos T y tras un período variable de tiempo un clon celular proliferaría de manera independiente de los mecanismos reguladores (23). Tuyp y cols. (24) y Whittemore y cols. (25) investigaron el papel de la estimulación antigénica crónica a muy diversas sustancias, incluidas potenciales carcinógenos, en pacientes con MF y grupos control; sus resultados fueron que en el grupo de pacientes con MF había mayor prevalencia de cánceres distintos a linfomas no Hodgkin, mayor tendencia a quemaduras solares y una historia familiar de atopia más frecuente, concluyendo finalmente que la exposición antigénica crónica a los químicos estudiados no era causa de MF.
— Genética. La ausencia de una agregación familiar bien documentada indica que la MF no es primariamente una enfermedad genética, aunque no se puede descartar cierto grado de predisposición a padecerla (17, 26). En algunos estudios se ha encontrado cierta asociación con determinados antígenos HLA, principalmente HLA-DR5 (27), y se han descrito anomalías numéricas o estructurales en algunos cromosomas (3, 28-30), principalmente en el cromosoma 1, donde además se sospecha que la región entre 1p22 y 1p36 podría contener un gen relacionado con el proceso de transformación y/o progresión de la MF (30). Recientemente Navas y cols. (31) sugirieron una relación entre hipermetilación del gen p16 (localizado en el brazo corto del cromosoma 9) y curso clínico desfavorable.
MANIFESTACIONES CLÍNICAS
Micosis fungoide
Las manifestaciones clínicas de la MF son muy variables. Se pueden agrupar en dos apartados:
— Micosis fungoide clásica (tipo Alibert-Bazin).
— Otras formas clinicopatológicas de micosis fungoide.
Micosis fungoide clásica (tipo Alibert-Bazin)
La MF clásica o de tipo Alibert-Bazin se divide en tres períodos o fases evolutivas denominadas: premicósica o eritematosa, en placas infiltradas y tumoral. Estas tres fases pueden superponerse entre ellas.
Fase premicósica o eritematosa
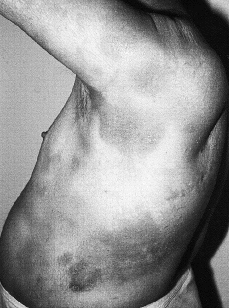
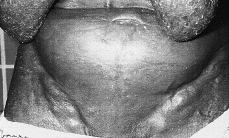
Las lesiones cutáneas son muy variadas. El tipo más común es el de placas de tamaño variable y formas generalmente redondeadas u ovales, eritematosas o eritematoescamosas, a veces con cierto tinte parduzco, poco o nada infiltradas al tacto, estables y de límites bien definidos o difusos e imprecisos (32-35) (Fig. 1). Se localizan con preferencia en tronco y raíz de miembros. En algunos casos las lesiones adoptan otras formas menos frecuentes como manchas con aspecto roseoliforme, placas psoriasiformes o erupciones papulosas e incluso vesiculosas, con aspecto de eccema (32), que hacen de la micosis fungoide una gran simuladora como en su tiempo se dijo de la sífilis (36). Un signo de ayuda diagnóstica es la persistencia de islotes reservados de piel sana en el centro de las placas (32).Todas estas lesiones tan diversas tienen en común el prurito intenso, que va provocando secundariamente excoriaciones, eccematización o infecciones secundarias; sin embargo, en algunos casos las lesiones son poco o nada pruriginosas (32). Esta primera fase puede durar varios años, e incluso décadas, aunque generalmente con el tiempo los elementos se vuelven más numerosos, se extienden y confluyen unos con otros. La duración media desde el inicio de los síntomas hasta el diagnóstico es de unos 6 años (37).
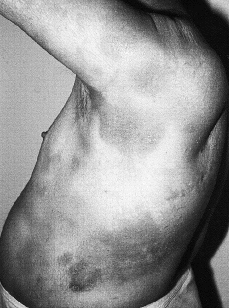
FIG. 1.--MF en fase premicósica. Placas eritematoescamosas, no infiltradas, localizadas en tronco y raíz de brazos.
La entidad conocida como parapsoriasis en grandes placas, caracterizada por placas eritematoescamosas de tamaño grande, a veces con áreas de atrofia y pigmentación (poiquilodermia atrófica vascular), es considerada por algunos autores como una MF en fase inicial (35, 38, 39).
Fase de placas infiltradas,
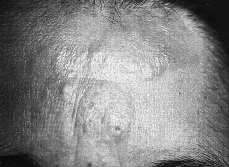
Esta segunda fase se instaura de forma paulatina con la aparición, sobre lesiones previas o en piel sana, de placas infiltradas. La infiltración aparece a menudo sobre el borde de ciertas placas, que incluso pueden llegar a formar figuras anulares o serpinginosas, con centro rosado, deprimido, finamente escamoso y borde sobrelevado de color rojo vivo. En otros casos la infiltración es más homogénea, apreciándose una placa bien circunscrita, abollonada y con borde en pendiente suave (Fig. 2). La superficie puede ser lisa o ligeramente escamosa, aunque a veces aparece erosionada y con costras (32).
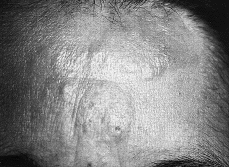
FIG. 2.--MF en fase de placas infiltradas. Placas elevadas, eritematosas y de bordes bien definidos, en pendiente suave.
Fase tumoral
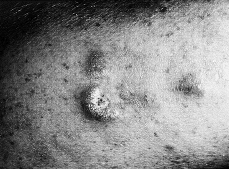
La fase tumoral se desarrolla gradualmente y con el tiempo, bien sea sobre piel sana o sobre piel previamente afecta, aparecen tumores que pueden ser hemisféricos o mostrar forma de hongo o seta, por constricción de la base (Fig. 3). Son de consistencia elástica o semiblanda. Su superficie es lisa y de coloración rojiza o purpúrica, aunque con frecuencia se ulceran o necrosan, dando lugar a úlceras más o menos profundas (32).
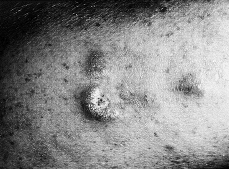
FIG. 3.--MF en fase tumoral. Tumores infiltrados y de coloración violácea.
Los casos descritos como MF tumoral d'emblée (forma tumoral desde el comienzo) (32, 37) se cree en la actualidad que no son MF tumorales, sino que corresponderían a linfomas T periféricos (35).
La afectación de mucosas es muy rara, aunque se han observado placas erosivas e infiltradas, e incluso tumores, en las mucosas bucofaríngea, ocular, digestiva, respiratoria y genital (32).
Otras formas clinicopatológicas de micosis fungoide
Además de la MF clásica tipo Alibert-Bazin podemos encontrar otras variantes menos frecuentes como son:
Micosis fungoide asociada a mucinosis folicular
La asociación entre MF y mucinosis folicular está ampliamente reconocida en la literatura, pudiendo esta última preceder, concurrir o aparecer posteriormente a la MF (40). La clasificación EORTC para los linfomas cutáneos primarios (1) la definió como una variante distintiva de MF caracterizada por infiltrados foliculotropos con degeneración mucinosa del folículo y que afecta preferentemente a cabeza y cuello. La clínica de estos casos es muy polimorfa y se han descrito distintas formas de expresión como alopecia no inflamatoria tipo alopecia areata; pápulas foliculares, que pueden confluir en placas de diferentes tamaños; placas eritematosas e infiltradas, nódulos y tumores, todo ello asociado con frecuencia a pérdida de pelo y prurito (1, 40). Recientemente, Bonta y cols. (41), en una revisión sobre este tipo de MF, encontraron que esta variante foliculotropa mostraba un curso más agresivo que la MF clásica.
MF folicular o quística
En algunos casos el infiltrado linfoide atípico se localiza exclusivamente a nivel peri o intrafolicular, sin epidermotropismo o mucinosis folicular. Este foliculotropismo da lugar a lesiones clínicas diferentes a las observadas en la MF clásica y que consisten en lesiones con apariencia de comedones o quistes miliares, pápulas foliculares, alopecia o placas con aspecto de piel de naranja (42-47).
Reticulosis pagetoide o enfermedad de Woringer-Kolopp
Se trata de una variante distintiva de MF caracterizada por la presencia de placas, generalmente únicas y localizadas principalmente en miembros, eritematoescamosas, psoriasiformes, con un borde moderadamente elevado y circinado, que a veces muestran un centro más claro o normal. Tiene un curso crónico y su evolución se considera benigna (1, 48-50). Existe una forma diseminada (tipo Ketron-Goodman) (51) que tiene una histopatología similar (52), pero el término de reticulosis pagetoide debe de ser reservado para el tipo localizado o enfermedad de Woringer-Kolopp (1). Se ha informado, de manera aislada, sobre la progresión del Woringer-Kolopp a la forma diseminada de reticulosis pagetoide (53), pero esto parece ser algo excepcional.
MF unilesional
Es una variante de MF caracterizada por lesiones cutáneas solitarias, en forma de placas eritematosas o eritematoescamosas (excepto un caso que presentó un nódulo), de varios centímetros de diámetro, localizadas principalmente en tronco o miembros superiores y de comportamiento generalmente benigno (54-57).
El diagnóstico diferencial con la enfermedad de Woringer-Kolopp puede ser difícil (54). Para Verret y cols. (56) la enfermedad de Woringer-Kolopp se diferencia de la MF unilesional por la clínica (aspecto más verrucoso, borde policíclico y tendencia a la curación central), histopatología (afectación principalmente epidérmica de linfocitos con citoplasma pálido y voluminoso) e inmunohistoquímica (el fenotipo de los linfocitos es con frecuencia CD8+). No obstante, Burns y cols. (58) consideran que el Woringer-Kolopp es una MF unilesional que muestra unas características histopatológicas, inmunofenotípicas y genotípicas similares a las fases iniciales de la MF, con un curso clínico prolongado y que requiere una estrecha vigilancia y un tratamiento conservador.
Micosis fungoide simulando dermatitis pigmentaria y purpúrica
En algunos casos la MF se ha presentado en forma de lesiones purpúricas en miembros inferiores, semejando una dermatitis purpúrica y pigmentaria (59-61). Según Lipsker y cols. (61) se debe de sospechar MF en aquellas dermatitis purpúricas y pigmentarias extensas, de más de 1 año de evolución, con patrón reticulado y patch test negativos. Las características histológicas que deben sugerirnos la presencia de MF son:
— Infiltrado polimorfo, con eosinófilos y células plasmáticas.
— Poca espongiosis y mayor grado de exocitosis (epidermotropismo).
— Mayor densidad de infiltrado en banda en dermis papilar.
— Presencia de linfocitos atípicos, cerebriformes, en el infiltrado (59).
MF eritrodérmica
En algunos casos, bien sobre lesiones previas de MF o de novo sobre piel sana (hombre rojo de Hallopeau), aparece un cuadro de eritrodermia (32, 35, 37, 62). No existe unanimidad respecto a si la eritrodermia como expresión de MF es o no diferente a la que aparece en el SS (35).
Otras formas
En la literatura dermatológica existen descripciones de distintas formas de presentación de la MF poco frecuentes, como, por ejemplo, hipopigmentada (63, 64), vesiculoampollosa (65), palmoplantar (66), pustular (67) e incluso la denominada como MF invisible, que se manifiesta en forma de prurito sin lesiones cutáneas (68). La entidad conocida como piel laxa granulomatosa es considerada, en la clasificación de la EORTC para linfomas cutáneos primarios, como un linfoma cutáneo de células T encuadrado en el apartado provisional (1), habiéndose descrito su asociación con MF clásica (69) y enfermedad de Hodgkin (70).
MANIFESTACIONES EXTRACUTANEAS
En las fases avanzadas la MF puede mostrar manifestaciones extracutáneas, principalmente linfadenopatías, y posteriormente afectación de órganos internos, como hígado, bazo, pulmón, tracto gastrointestinal y médula ósea (34). Con frecuencia las lesiones viscerales son asintomáticas o infravaloradas, ante lo llamativo del cuadro cutáneo, y se trata en muchos casos de hallazgos de autopsia (35). En general, la afectación visceral se observa en pacientes con lesiones cutáneas avanzadas, adenopatías y células de Sézary circulantes (33).
A menudo la causa de muerte en los pacientes con MF, principalmente en fase de placas localizadas, es debida a alguna enfermedad interrecurrente de origen cardiovascular u otra neoplasia. En un estudio realizado en Stanford sobre 465 pacientes fallecieron 245, y de éstos en el 47% la causa de muerte fue debida a una enfermedad intercurrente; sin embargo, la mayoría de los casos que desarrollaron tumores o eritrodermia fallecieron por la MF (37).
Síndrome de Sézary
El SS se define por la tríada de eritrodermia, linfadenopatías generalizadas y presencia de células T neoplásicas en piel, ganglios linfáticos y sangre periférica (1, 4). No está aún bien determinado el número de células atípicas circulantes que son necesarias para definir el síndrome. Winkelmann y cols. (71) propusieron la cifra de más de 1.000 células atípicas circulantes mm3 para definir el síndrome, de manera que aquellos pacientes con menos de 1.000 corresponderían a casos de pre-Sézary. El Grupo de Estudio del Linfoma Cutáneo de la EORTC (1) sugirió como criterio adicional la demostración en sangre periférica de células T clonales y la presencia de una población elevada de células T CD4+, con el consiguiente incremento de la relación CD4/CD8 (> 10).
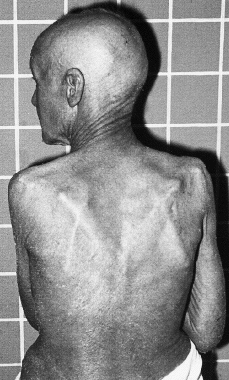
Clínicamente se manifiesta en forma de eritrodermia y adenopatías, con edema e intenso prurito (Figs. 4 y 5). Adicionalmente se puede encontrar hepatomegalia (57% de los casos), alopecia (32%), onicodistrofia (32%), queratodermia (29%), ectropión con inflamación ocular secundaria y en las formas severas facies leonina (72).
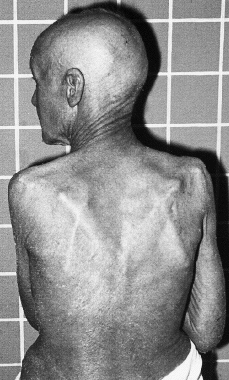
FIG. 4.--SS. Eritrodermia y alopecia en cuero cabelludo.
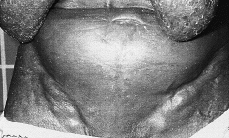
FIG. 5.--SS. Adenopatías voluminosas en regiones inguinales.
Aunque el SS presenta una personalidad clínica propia, diferente de la MF clásica, tradicionalmente se ha presentado junto con la MF como prototipo de linfoma cutáneo de células T epidermotropo. No obstante, la reciente clasificación de los linfomas cutáneos primarios propuesta por la EORTC incluyó al SS en el grupo de los linfomas cutáneos primarios agresivos, con una supervivencia a los 5 años del 11% (1).
HISTOPATOLOGÍA
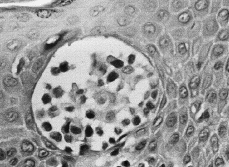
En las fases iniciales de la MF el diagnóstico histológico puede ser difícil. Se caracteriza por un infiltrado linfocitario escaso en dermis papilar, con tendencia a disponerse en banda subepidérmica (Fig. 6). Estos linfocitos presentan epidermotropismo o tendencia a invadir la epidermis. Generalmente aparecen como linfocitos aislados, rodeados de un halo claro, y localizados principalmente en las capas inferiores de la epidermis, con mínima o nula espongiosis; a veces se distribuyen de una manera lineal a lo largo de la unión dermoepidérmica en una disposición como «en fila» (73). La presencia de acúmulos o grupos de linfocitos en la epidermis separados de los queratinocitos adyacentes por un halo claro (microabscesos de Pautrier) es un criterio clásico en el diagnóstico de la MF, pero en las fases iniciales aparece en menos de un tercio aproximado de los casos (74, 75) (Fig. 7).

FIG. 6.--Infiltrado linfocitario ligero en dermis papilar, con tendencia a disponerse en banda.
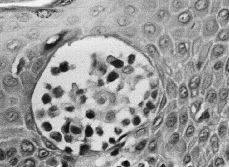
FIG. 7.--Microabsceso de Pautrier.
Son también importantes para el diagnóstico las alteraciones arquitecturales que afectan a la dermis papilar y a la epidermis. Éstas consisten en un ensanchamiento y fibrosis de la dermis papilar, cuyos haces de colágeno aparecen engrosados y dispuestos horizontalmente a la superficie epidérmica, en contraste con la disposición vertical que se observa en la liquenificación (73, 75). La epidermis suele mostrar ligera hiperplasia psoriasiforme e hiperqueratosis, aunque en las formas de poiquilodermia aparece atrofia (73).
La atipia de los linfocitos puede ser difícil de demostrar, pero siempre se debe de intentar identificar células de núcleos hipercromáticos o cerebriformes. A veces es más fácil encontrarles en la epidermis que en la dermis, y esto es debido a que virtualmente todos los linfocitos intraepidérmicos son neoplásicos, mientras que de los linfocitos localizados en la dermis papilar sólo una parte son neoplásicos (73, 74).
A medida que la enfermedad progresa y las placas se van engrosando, el infiltrado se hace mayor y se dispone en banda densa en dermis papilar y a nivel de dermis reticular, adopta un patrón perivascular superficial y profundo o casi difuso (73) (Fig. 8). Estos linfocitos son más atípicos, de mayor tamaño y con frecuencia forman en la epidermis microabscesos de Pautrier.

FIG. 8.--Infiltrado en banda densa subepidérmica.
Finalmente, en la fase tumoral disminuye el grado de epidermotropismo y el infiltrado tiende a hacerse más denso y profundo (73, 74). En esta fase algunas células pierden la morfología cerebriforme característica y se transforman en células grandes. Para Salhany y cols. (76 ) cuando estas células transformadas exceden el 25% del infiltrado o forman nódulos microscópicos se considera que la MF ha sufrido una transformación a linfoma de célula grande.
En el SS se observa habitualmente un infiltrado en banda subepidérmica de linfocitos, con grados variables de atipia y epidermotropismo (73, 77-79); no obstante, Buechner y cols. (77) hallaron que el 17% de las biopsias de piel de pacientes con SS mostraban solamente cambios de dermatitis crónica con un infiltrado perivascular moderado de linfocitos pequeños sin atipia, histiocitos y algún eosinófilo.
INMUNOHISTOQUÍMICA
Desde el punto de vista inmunohistoquímico la célula proliferante en la MF/SS es el linfocito T maduro cooperador de memoria (CD3+, CD5+, CD43+, CD45RO+, CD45+, CD4+, CD8, CD20, CD30)tivo, a diferencia de las células T maduras (82). Se han descrito casos aislados con fenotipo T supresor CD4 CD8+ (83) y en las fases tardías de la MF, principalmente tumoral, puede haber inmunofenotipos aberrantes (84).
DIAGNÓSTICO
El diagnóstico de la MF/SS se basa en los hallazgos clínicos combinados con la histología de la biopsia de piel (85). En aquellos pacientes con lesiones incipientes los hallazgos histológicos pueden ser poco específicos y en estos casos es importante realizar un seguimiento periódico del enfermo y practicar biopsias repetidas. Existen, no obstante, técnicas diagnósticas adicionales que pueden ser útiles en determinados casos o ayudar al diagnóstico precoz. Algunas de estas técnicas, como la microscopia electrónica, citometría del DNA e índice del contorno nuclear, apenas se utilizan en la práctica clínica cotidiana y han sido desplazadas en los últimos años por el diagnóstico inmunogenético.
El diagnóstico inmunogenético de la MF/SS puede realizarse con técnicas de Southern blot o de PCR (reacción en cadena de la polimerasa).
— Técnicas de Southern blot. Con esta técnica se estudia el gen que codifica la cadena beta del receptor T. Presenta algunas limitaciones como la utilización de tejido fresco o congelado y una sensibilidad limitada (sólo permite detectar una proliferación clonal cuando ésta representa entre el 1-5% del DNA total), por lo que no es útil para detectar monoclonalidad en los primeros estadios de la MF en los que el porcentaje de células neoplásicas en relación con el infiltrado benigno reactivo es mínimo (80).
— Reacción en cadena de la polimerasa (PCR). En la actualidad la PCR está desplazando al Shouthern blot por su mayor sencillez, sensibilidad y permitirnos manejar material incluido en parafina. En este caso se estudia el gen que codifica la cadena gamma del receptor T (80, 85). Con la PCR se ha encontrado reordenamiento clonal en la mayoría de los casos de MF/SS, incluidas las fases iniciales (86-89), demostrando mayor sensibilidad con respecto al Southern blot; no obstante, se insiste en que monoclonalidad no es sinónimo de malignidad y estos datos deben de ser interpretados junto a la clínica e histopatología (3).
ESTUDIO DE EXTENSIÓN Y CLASIFICACIÓN
Una vez realizado el diagnóstico de MF/SS se procede a practicar un estudio de extensión de la enfermedad. En nuestro Servicio se emplea un protocolo de estudio que incluye la solicitud a todos los pacientes de hemograma y fórmula leucocitaria, con determinación de células de Sézary circulantes, VSG, bioquímica hemática, enzima láctico deshidrogenasa (LDH), beta 2 microglobulina, radiografía de tórax y biopsia de piel. La mayoría de los investigadores están de acuerdo en hacer biopsia de ganglios linfáticos sólo en aquellos casos en que éstos están agrandados (90). El valor de la biopsia de médula ósea o el escáner abdominal y pélvico está menos determinado y debe de ser reservado para aquellos pacientes con tumores, linfocitos atípicos circulantes, afectación ganglionar o síntomas sugestivos (90).
Conocida la extensión de la enfermedad se clasifica a los pacientes en distintos estadios. El sistema de clasificación más empleado es el sistema TNM, introducido en 1979 por el National Cancer Institute (91) (tabla II) y modificado en 1988 por Sausville y cols. (92) (tabla III).


FACTORES PRONÓSTICOS
En la actualidad se aceptan como factores pronósticos bien establecidos la extensión y tipo de lesión cutánea, la afectación ganglionar y la invasión de órganos internos y sangre periférica (92-103). Sausville y cols. (92) en 1988 establecieron tres grupos pronósticos:
— Grupo de bajo riesgo, con supervivencia mediana de más de 12 años. Comprendía aquellos pacientes con lesiones cutáneas en forma de placas cutáneas (T1, T2), con histología ganglionar LN1, LN2, y sin afectación de sangre periférica o vísceras.
— Grupo de alto riesgo, con supervivencia mediana menor de 3 años. En aquellos casos con afectación visceral, confirmada histológicamente, o cuando existía borramiento completo de la estructura ganglionar.
— Grupo de riesgo intermedio, con supervivencia mediana de 5 años. Comprendía el resto de los casos.
Existen, no obstante, otros parámetros estudiados en los últimos años y que se han comportado como buenos indicadores pronósticos. Entre ellos destacamos algunos parámetros analíticos como el valor sérico de la lactato deshidrogenasa (LDH) (98, 102), beta 2 microglobulina (102) y el receptor soluble de la interleucina 2 (sIL-2R) (104); parámetros histopatológicos, principalmente la medida del grosor del infiltrado cutáneo (> 1 mm) (98), y parámetros inmunohistoquímicos como la correlación existente entre el porcentaje alto de CD8 y mejor supervivencia (105).
Es probable que el diagnóstico y tratamiento precoz, particularmente en las fases iniciales, pueda mejorar la supervivencia. En este sentido resaltamos los trabajos de Kim y cols. (106) realizados con 122 pacientes en estadio IA y de Zackheim y cols. (100) en 174 pacientes en estadio T1; ambos hallaron que la supervivencia a largo plazo era similar a la esperada en una población control.
En el SS la supervivencia es menor y se ha estimado entre 2,5-5 años (72). Kim y cols. (107) realizaron un estudio retrospectivo sobre los factores pronósticos en 106 pacientes con MF eritrodérmica y SS y establecieron tres grupos pronósticos:
— Grupo favorable, con supervivencia mediana de 10,2 años. Incluía aquellos pacientes con edad menor de 65 años, estadio clínico III y ausencia de afectación de sangre periférica.
— Grupo desfavorable, con supervivencia mediana de 1,5 años. Estaban aquellos casos con edad mayor de 65 años, estadio clínico IV y afectación de sangre periférica.
— Grupo intermedio, con supervivencia mediana de 3,7 años. Comprendía el resto de pacientes.
TRANSFORMACIÓN A LINFOMA DE CÉLULA GRANDE (LCG)
Algunos pacientes con MF/SS desarrollan a lo largo de su evolución un cambio en la morfología celular, con transformación de las células de pequeño o mediano tamaño en células grandes (76, 108-111). En general se acepta que cuando las células grandes constituyen más del 25% del infiltrado o bien forman nódulos microscópicos, la MF/SS se ha transformado a LCG (76). La transformación a LCG aparece con una frecuencia variable, dependiendo de los criterios que se utilicen para definirla y de la selección de pacientes, oscilando entre el 8-55% de los casos (76, 108-111). Con frecuencia se acompaña de una evolución más agresiva de la enfermedad y supervivencia mediana más corta (76, 108-110).
TRATAMIENTO
En la actualidad no existe un único tratamiento para la MF, sino que nos encontramos ante distintas alternativas terapéuticas cuyo objetivo principal es ofrecer un aumento en las expectativas de vida, así como una mejora de su calidad. El tratamiento actual de la MF incluye, principalmente, las siguientes posibilidades:
Corticoides tópicos
Pueden ser útiles en fases iniciales (112), aunque generalmente la respuesta es incompleta y de corta duración (113).
Quimioterapia tópica
La quimioterapia tópica está indicada principalmente para el tratamiento de la MF en placas no infiltradas o que presentan una infiltración ligera o moderada (113). Disponemos de dos sustancias: mecloretamina (actualmente no comercializada) y carmustina.
— Hidrocloruro de mecloretamina (mostaza nitrogenada). Se puede usar en solución acuosa a una concentración de 10 mg/60 ml o en pomada a una concentración de 10 mg/100 g (113). Se aplica diariamente por toda la superficie cutánea (exceptuando genitales y pliegues) hasta la regresión de las lesiones, que ocurre generalmente entre 3-6 meses para la solución y 6-12 meses para la pomada (114). Aunque se recomienda el tratamiento sobre toda la superficie cutánea, la necesidad de aplicarlo en áreas no afectas es controvertida (114). La tasa de respuestas completas varía entre el 50-75% para el estadio T1 y del 25-50% para T2 (114). Con frecuencia ocasiona irritación o sensibilización, e incluso puede dar lugar a carcinomas cutáneos (114, 115).
— Carmustina tópica (BCNU). Se utiliza en forma de solución alcohólica mezclada con agua (solución) o con vaselina (pomada) y se aplica una vez al día por toda la superficie cutánea (respetando cara, manos, genitales y pliegues) o sólo en áreas afectas (114-116). Con la solución se ha obtenido un 86% de respuestas completas en pacientes con estadio T1 y un 48% para los casos con estadio T2 (114). La mayoría de los pacientes experimentan algún grado de eritema que, a veces, es seguido de telangiestasias. No se han descrito cánceres cutáneos y la depresión de médula ósea es un efecto secundario poco frecuente (85, 114).
Ultravioleta B
Se ha encontrado buena respuesta en fases iniciales, tanto con la modalidad de banda ancha (117) como de banda estrecha (311 nm) (118); sin embargo, no se recomienda en placas infiltradas debido a la penetración limitada de los UVB (114).
Fotoquimioterapia (PUVA)
Es uno de los tratamientos de elección en los primeros estadios, consiguiéndose respuestas completas en el 90% de los pacientes con estadio IA, 76% en IB y 78% en IIA (119). La utilidad del PUVA se limita principalmente a la forma de placas cutáneas (IA a IIA), ya que en el estadio tumoral (IIB) tiene sólo efecto paliativo por la penetración limitada de los UVA (120) y en los pacientes con eritrodermia (estadio lll) el porcentaje de recidivas es elevado (119, 120).
Retinoides orales
Son derivados de la vitamina A, cuya actividad en la MF sería debida a su efecto antitumoral e inmunomodulador (85, 120). Tanto el etretinato como la isotretinoína son igual de eficaces (121). Son útiles principalmente en los estadios precoces de la MF, con una respuesta media del 60% (120); sin embargo, la duración de la remisión suele ser breve (120, 121), por lo que no son recomendados como tratamiento de primera elección, a no ser que se asocien a otras modalidades como, por ejemplo, PUVA.
Retinoides orales más PUVA (RePUVA)
Se han logrado respuestas completas en un 73% de los casos, cifra similar a la obtenida con PUVA; sin embargo, con este método se administran menos sesiones de PUVA y nos permite prolongar las remisiones administrando los retinoides como terapia de mantenimiento (122).
Fotoquimioterapia extracorpórea
Combina aspectos de la leucoféresis convencional y la fototerapia con 8-metoxipsoraleno y radiación ultravioleta A. Los mejores resultados se han conseguido en la MF eritrodérmica (114, 123).
Interferón alfa
Es un modificador de la respuesta biológica que se ha mostrado eficaz en el tratamiento de los linfomas cutáneos. Se han conseguido respuestas hasta en un 64% de los casos de MF, tanto en fases iniciales como tardías (124); sin embargo, la tasa de respuestas completas es baja, entre el 16-18% (120). Existe una considerable variación en las dosis y esquemas de tratamiento, aunque generalmente se comienza por dosis bajas, de 3 millones de UI tres veces a la semana y se aumenta durante un período variable de inducción en función de la tolerancia del paciente hasta 9-18 millones de UI (113, 114). En algunos casos se ha empleado de forma intralesional en placas cutáneas de MF, logrando remisión completa en un porcentaje alto de las lesiones (125).
El interferón también se ha utilizado combinado a otros agentes, principalmente PUVA (126, 127), donde se obtuvieron respuestas completas incluso en pacientes que no respondieron inicialmente a PUVA solo (127).
Baño de electrones (electron beam therapy)
Algunos autores como Zackheim y cols. (114) lo consideran el tratamiento de primera elección para aquellos pacientes con placas infiltradas diseminadas o tumores. Con esta técnica en la Universidad de Stanford obtuvieron respuestas completas en el 98% de placas localizadas, 71% de placas generalizadas y 36% de tumores, aunque en la mayoría de los casos las lesiones rebrotaron en los 5 primeros años (128). Para evitar este número elevado de recidivas se ha propuesto un tratamiento de mantenimiento con mecloretamina tópica (129) o PUVA (130), si bien esto puede incrementar el riesgo de carcinomas cutáneos (1 31).
Radioterapia convencional de ortovoltaje
Puede ser efectiva para erradicar tumores cutáneos aislados (113).
Quimioterapia sistémica
Se ha usado el metotrexate a dosis bajas (solo o en combinación con corticoides tópicos) en aquellos pacientes con MF en placas que no responden adecuadamente a PUVA o quimioterapia tópica (113). Para Zackheim y cols. (114) el metrotrexate sería el tratamiento de primera elección en las fases iniciales o intermedias de la MF eritrodérmica.
En los estadios IVA y IVB se utilizan diversas asociaciones de drogas como adriamicina, vincristina, ciclofosfamida, prednisona, etc. Una combinación frecuente es: ciclofosfamida, adriamicina, vincristina y prednisona (CHOP) (114, 132).
Diversas combinaciones entre los tratamientos mencionados (120, 133-135)
A modo de esquema general se puede seguir la siguiente pauta:
— Para los estadios IA, IB y IIA se utiliza PUVA o quimioterapia tópica como tratamiento de primera elección.
— En el estadio IIB se recomienda baño de electrones y a continuación, de manera opcional o para lesiones refractarias al tratamiento, aplicar quimioterapia tópica o PUVA.
— En el estadio III fotoquimioterapia extracorpórea o metotrexate.
— En estadios avanzados, IVA y IVB, es necesario quimioterapia sistémica.
En el SS se ha utilizado la llamada pauta d Winkelmann, que consiste en administrar 20 mg/día de prednisona, en dosis única por la mañana, junto con 4 mg/día de clorambucil (136). El metotrexate a dosis preferentemente bajas, entre 10-50 mg por semana, es una alternativa eficaz en muchos casos (137). En la actualidad no está aclarado el beneficio de la fotoféresis extracorpórea con respecto a los tratamientos convencionales en el SS (138).


