





INTRODUCCION
El eritema elevatum et diutinum (EED), o eritema elevado y persistente, fue descrito en 1894 por Radcliffe Crocker1. Si bien en los trabajos iniciales se diferenciaba un «tipo Bury», en mujeres jóvenes con historia de enfermedad reumática, de un «tipo Hutchinson», que afectaba a varones ancianos2, hoy se considera una única entidad con independencia de las posibles causas subyacentes3. Se trata de una enfermedad cutánea crónica que forma parte del espectro de las vasculitis leucocitoclásticas cutáneas con una fase de involución fibrótica muy prolongada4,5. Se ha señalado la curiosa discrepancia entre el curso crónico de la enfermedad y el hallazgo histológico de vasculitis aguda6. A pesar del curso prolongado, a veces durante años, hay una notable ausencia de afectación sistémica por el proceso vasculítico.
Las habituales lesiones elementales que aparecen en el EED son pápulas y placas induradas, si bien en ocasiones se observa también progresión hacia ampollas o úlceras. El color de cada elemento es rojo intenso, violáceo o marrón. Las lesiones suelen localizarse de manera bilateral y simétrica en la piel suprayacente a las articulaciones de las superficies de extensión de las extremidades, especialmente codos, dorso de manos, rodillas, piel sobre el tendón de Aquiles y en ocasiones, nalgas o cara. A la palpación la consistencia de cada lesión aumenta con el paso del tiempo. La involución parcial puede darles un color amarillo semejante al de los xantomas. En ocasiones el EED puede resultar un simulador clínico de otras patologías como el sarcoma de Kaposi7, la angiomatosis bacilar8, queloides9 o xantomas3.
Existe correlación entre la edad de las lesiones de EED y los hallazgos histopatológicos predominantes10. El estadio inicial de la enfermedad se caracteriza por una vasculitis franca, definida por la existencia de necrosis fibrinoide de la pared vascular, junto a la cual las células inflamatorias predominantes son neutrófilos con leucocitoclasia. Puede existir edema de la dermis papilar cuyo correlato clínico es un aspecto de pseudovesiculación. En lesiones de mayor antigüedad el infiltrado inflamatorio va siendo reemplazado por tejido de granulación, una importante fibrosis e histiocitos xantomatizados por deposición secundaria de material lipídico11. Este último fenómeno fue responsable del término colesterolosis extracelular con el que se denominó a este proceso12; sin embargo, la antigua nomenclatura se ha demostrado inadecuada ya que en los depósitos lipídicos también hay fosfolípidos y su localización es predominantemente intracelular13. En los elementos más evolucionados de EED predomina la fibrosis sobre el infiltrado celular: la dermis aparece dividida en compartimentos constituidos por gruesas bandas fibrosas concéntricas en un entramado de reticulina10. Las técnicas de inmunofluorescencia directa suelen resultar negativas, si bien en observaciones aisladas se ha descrito positividad para inmunoglobulinas, complemento y fibrina en las paredes vasculares4.
A continuación describimos una paciente con EED y peculiares hallazgos clínicos y analíticos asociados.
DESCRIPCION DEL CASO
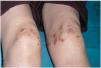
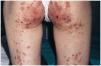
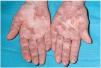
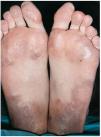
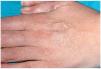
Una mujer de 24 años, sin otros antecedentes patológicos de interés, consultó por una dermatosis crónica persistente a lo largo de dos años que cursaba de manera asintomática. La exploración reveló la presencia de múltiples pápulas y placas induradas, de color violáceo a marrón, localizadas de manera bilateral y simétrica sobre la superficie de extensión de codos, rodillas (fig. 1), tobillos, así como en región glútea y cara posterior de muslos (fig. 2). Varias de las lesiones localizadas en la región glútea aparecían ulceradas. Presentaba, además, múltiples placas elevadas, firmes, confluentes, de apariencia pseudovesicular y pustulosa, que afectaban por completo la superficie palmar de las manos (fig. 3). También había afectación de la zona anterior y posterior de las plantas de los pies por placas violáceas de contorno figurado (fig. 4); la bóveda plantar estaba respetada. Entre las lesiones activas se podían encontrar cicatrices lenticulares atróficas («en papel de fumar») dejadas por la involución de algunas lesiones previas y además, en dorso de mano izquierda (fig. 5) y sobre maléolo externo derecho, otras cicatrices de mayor tamaño y con un característico aspecto cribiforme.
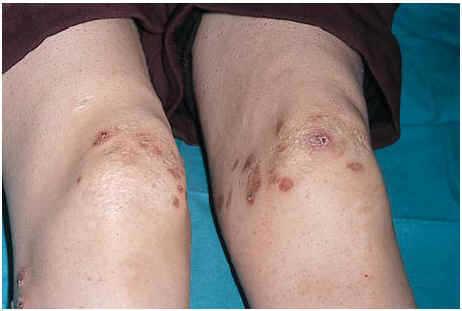
Fig. 1.--Placas violáceas sobre ambas rodillas.
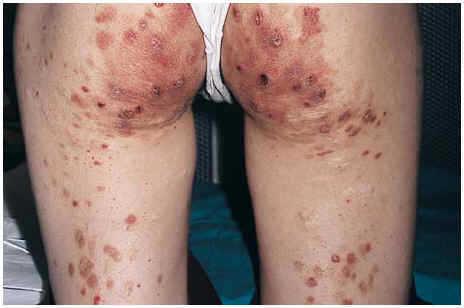
Fig. 2.--Lesiones ulceradas en región glútea.
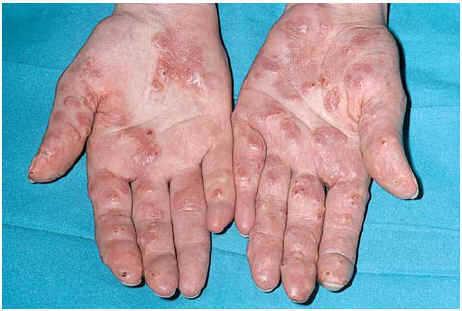
Fig. 3.--Intensa afectación palmar.

Fig. 4.--Afectación plantar.
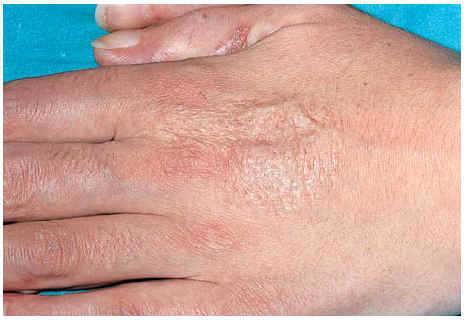
Fig. 5.--Cicatriz cribiforme en dorso de mano.
La biopsia de una lesión reciente, localizada en muslo izquierdo, reveló un infiltrado neutrofílico masivo y polvillo nuclear ocupando difusamente toda la dermis desde el cuerpo papilar hasta la hipodermis (fig. 6). En el seno del infiltrado neutrofílico se podían encontrar algunos vasos con necrosis fibrinoide de la pared (fig. 7). La inmunofluorescencia directa fue negativa. Los cultivos de una porción de biopsia para bacterias, hongos y micobacterias no revelaron crecimiento patógeno.
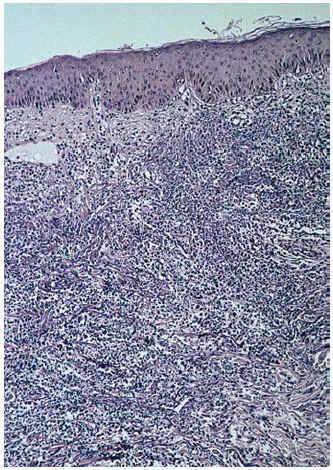
Fig. 6.--Aspecto histológico general.
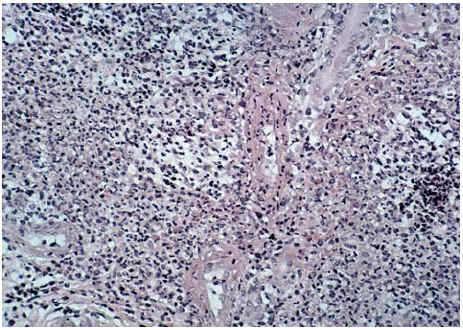
Fig. 7.--Vaso con necrosis fibrinoide de su pared.
Los estudios analíticos complementarios resultaron con valores dentro de la normalidad; la bioquímica sérica, función renal y hepática, inmunoelectroforesis de proteínas séricas, factor reumatoide, velocidad de sedimentación globular (VSG), ASLO, PCR, anticuerpos antinucleares (ANA), anti-ADN, SSA, SSB, RNP, C3, C4, anticuerpos antiendomisio, así como las serologías para virus de la hepatitis A (VHA), B (VHB) y C (VHC), lúes y virus de la inmunodeficiencia humana (VIH). Existía cierta anemia ferropénica (hematíes 3.600.000/mm3; hemoglobina 10 g/dl; hierro 38 µg/dl). Los anticuerpos anticitoplasma de neutrófilo (ANCA), determinados mediante técnica de inmunofluorescencia indirecta, resultaron positivos a título 1/160, con patrón de p-ANCA; la especificidad antigénica de estos autoanticuerpos del tipo IgG, analizada por inmunoanálisis (ELISA), estaba dirigida contra mieloperoxidasa (MPO) (niveles de anti-MPO: 59,5 U/ml; intervalo: positivo > 3 U/ml).
El tratamiento con sulfona 100 mg/día condujo a una respuesta clínica espectacular con evolución de todas las lesiones hacia la completa cicatrización tras sólo tres semanas de tratamiento. Se ha realizado una progresiva reducción de la dosis de sulfona. En la actualidad, un año después del inicio del tratamiento y con una dosis de 25 mg/día de sulfona, la paciente se mantiene en seguimiento clínico y analítico sin que hasta el momento haya experimentado reactivación de la enfermedad ni desarrollo de ninguna patología sistémica relacionada.
DISCUSION
La presencia de lesiones palmares en enfermos de EED ha sido referida en muy pocas ocasiones y a propósito, únicamente, de elementos escasos y aislados14-16. En ningún trabajo se describe una afectación palmar tan intensa como la que nuestra paciente presentaba. Se ha observado ocasionalmente afectación palmar pustulosa en otras dermatosis neutrofílicas como el síndrome de Sweet17. Por el contrario, las lesiones plantares o en el dorso de las manos no son infrecuentes en el EED. Recientemente se ha individualizado la denominada vasculitis pustulosa de las manos18, o dermatosis neutrofílica del dorso de las manos19, en función de la peculiar distribución de sus lesiones en la zona radial del dorso de las mismas, y es aún motivo de controversia la posible naturaleza primaria o secundaria de la vasculitis subyacente19, 20.
Es posible encontrar otras eventuales peculiaridades clínicas en enfermos con EED. La existencia de masas nodulares de gran tamaño se ha considerado sugestiva de asociación clínica con la infección por el virus de la inmunodeficiencia humana (VIH)16, 21. La presencia de lesiones ulceradas se puede interpretar bien como algo intrínseco al EED o bien como elementos de carácter intermedio entre EED y pioderma gangrenoso, para los cuales existirá una análoga evolución hacia la curación con peculiares cicatrices cribiformes como las que encontramos en nuestra paciente. Así, el EED se considera una de las dermatosis neutrofílicas, grupo de enfermedades que comparten numerosas características22 y de las que hay ejemplos de formas intermedias o complejas de difícil clasificación23. Para el EED se ha referido coexistencia o superposición con piodermia gangrenoso24 o síndrome de Sweet25, de los que se distingue, esencialmente, por la presencia constante de vasculitis11.
Existe un paralelismo clínico e histológico entre el EED y el granuloma facial que además puede presentarse con afectación extrafacial26, 27 o diseminada28. Con una visión generalizadora algunos autores29 han denominado «venulitis crónica fibrosante localizada» a un conjunto clínicamente diverso que comparte un cuadro histológico consistente en una vasculitis leucocitoclástica cutánea crónica localizada que evoluciona hacia un patrón de fibrosis estoriforme o concéntrica (angiocéntrica o «targetoide») acompañada por un infiltrado inflamatorio mixto. Dentro de este espectro se incluiría no sólo el EED (con predominio de neutrófilos y afectación de la dermis adventicial) y el granuloma facial (con predominio de eosinófilos y respeto de una zona Grenz), sino también algunos de los denominados pseudotumores inflamatorios (ricos en células plasmáticas)29, 30.
El EED se asocia, con frecuencia, a diversos problemas médicos, especialmente anomalías hematológicas. La asociación con gammapatía o mieloma del tipo IgA parece algo más que un hallazgo casual: en alguna serie4 el 31% de los pacientes con EED tenía además una gammapatía monoclonal IgA de tipo esencial. Si bien otras dermatosis neutrofílicas comparten esta asociación, el vínculo con el EED es más fuerte. Otras afecciones descritas en asociación con el EED se resumen en la tabla 1. La variedad de enfermedades asociadas podría tener como mecanismo patogénico común, para dar origen al EED, diversas situaciones en que con facilidad se produce la formación de inmunocomplejos, ya sean condiciones de exceso de exposición a antígenos (infecciones crónicas, enfermedad inflamatoria intestinal), como situaciones en que circulan altos niveles de anticuerpos (paraproteinemias, enfermedades autoinmunes)38. En muchos casos el EED precede al diagnóstico de las enfermedades asociadas.

Los autoanticuerpos dirigidos contra proteínas constituyentes del citoplasma de los neutrófilos (ANCA) parecen tener significado patogénico en un subgrupo de vasculitis sistémicas de vasos de pequeño tamaño, carentes de depósitos inmunes en el estudio por inmunofluorescencia directa, que incluye la granulomatosis de Wegener, el síndrome de Churg-Strauss y la poliarteritis microscópica42. También pueden encontrarse ANCA en enfermedades muy heterogéneas, entre las que se incluye el síndrome de Sweet43, la piodermia gangrenosa44 u otras dermatosis neutrofílicas complejas45, en la mayoría de las cuales es probable que los ANCA sólo sean un epifenómeno de reconocimiento de proteínas expuestas por la fragmentación de los neutrófilos. Los p-ANCA tienen como diana molecular la MPO, una proteína localizada en los gránulos azurófilos de neutrófilos, y se considera el patrón de ANCA más característico de la poliarteritis microscópica. El valor diagnóstico de los anticuerpos p-ANCA (anti-MPO) en las vasculitis está, sin embargo, menos establecido que el que se atribuye a los c-ANCA (antiproteinasa 3). Los ANCA son predominantemente del tipo IgG, si bien se han comunicado ANCA IgA en alguna serie de vasculitis en la que curiosamente se incluía un ejemplo de EED46.
Hemos descrito un ejemplo de EED con la peculiaridad clínica de la intensa afectación palmar, sin asociación demostrada hasta el momento, a otra patología sistémica y que respondió de manera espectacular al tratamiento con sulfona. La presencia de ANCA con patrón de inmunofluorescencia perinuclear en una paciente con EED, enfermedad cuyo sustrato patológico es una vasculitis de vasos pequeños, es un hallazgo interesante y que merece ser examinado en series amplias de enfermos.


