



Una mujer peruana de 49 años de edad presentaba linfadenopatía cervical y dolor de cuello de cinco meses de evolución. Los antecedentes sociales, familiares y médicos no aportaron nada significativo. En la exploración física, se evidenciaron adenopatías cervicales bilaterales y una placa eritematosa de 3 cm de diámetro en cuero cabelludo (fig. 1a). En la analítica destacaba neutropenia (2,2 x 109/l) y positividad para los anticuerpos antinucleares (títulos 1:160)
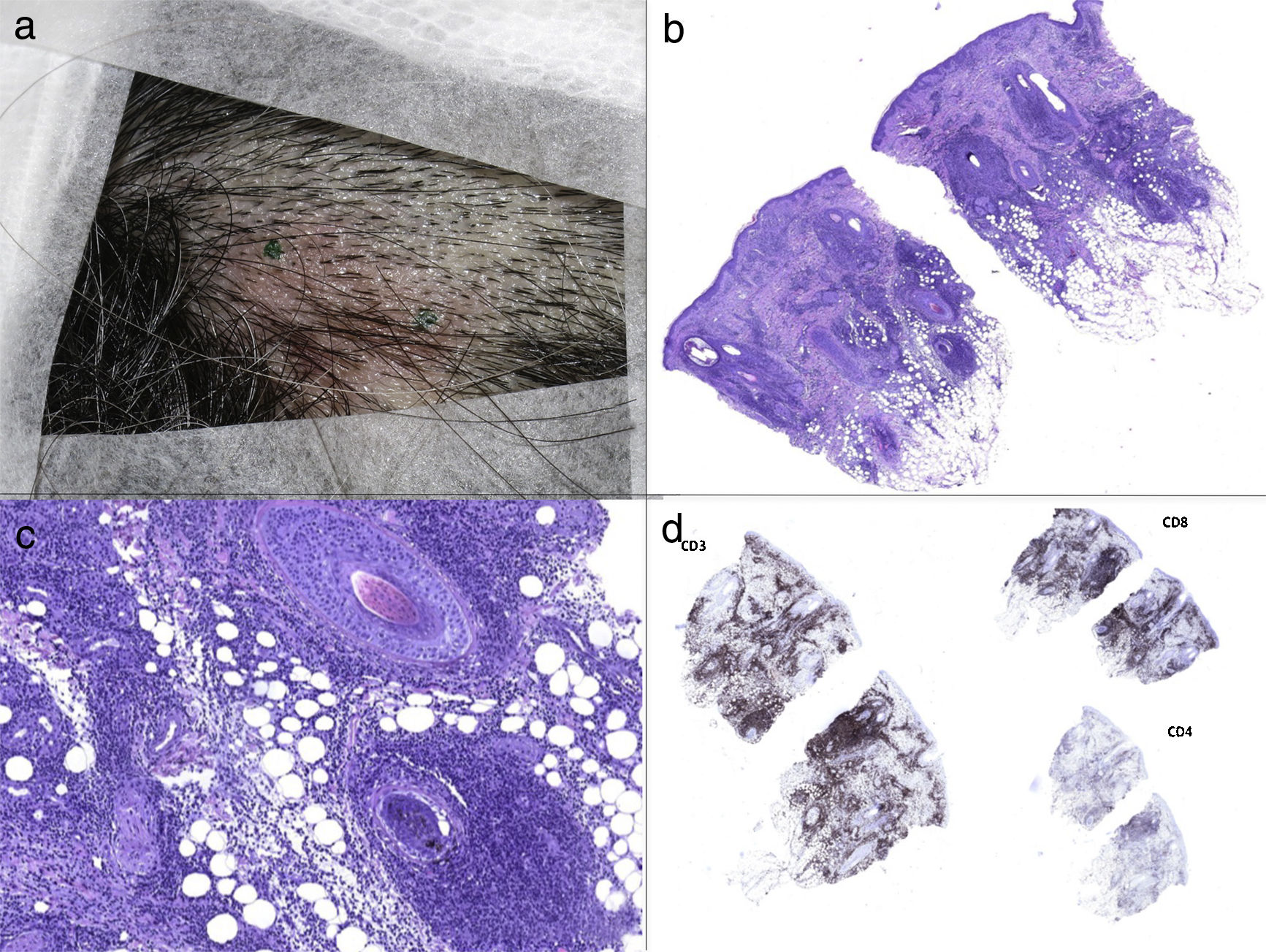
a) Placa eritematosa de 3cm de diámetro sobre cuero cabelludo. b) La tinción de hematoxilina-eosina reveló un cambio vacuolar en las células basales epidérmicas e infiltración linfohistiocítica perifolicular y cariorrexis en la dermis reticular (40x). c) Infiltración linfohistiocitaria perifolicular en tinción de hematoxilina-eosina (200x). d) El análisis inmunohistoquímico reveló que el infiltrado linfoide fue predominantemente CD3+, con células CD8 positivas predominantes sobre CD4.
Tanto la tomografía computarizada como la tomografía por emisión de positrones con 18 fluorodeoxiglucosa confirmaron la presencia de adenopatías cervicales bilaterales con ganglios linfáticos agrandados de hasta 2cm de tamaño.
La biopsia de piel mostró un leve cambio vacuolar en las células basales epidérmicas, acompañado de un infiltrado linfohistiocitario perifolicular junto con cariorrexis en la dermis reticular (fig. 1 b, 1c). El estudio inmunohistoquímico reveló que el infiltrado linfoide era CD3+, predominantemente CD8+ (fig. 1d). La positividad de CD68, CD163 y mieloperoxidasa puso en evidencia la presencia de múltiples histiocitos, y la positividad de CD123, la presencia de algunos monocitos plasmocitoides. No se detectaron neutrofilos, eosinófilos ni granulomas en el infiltrado, ni se encontraron hallazgos sugestivos de vasculitis
La biopsia de los ganglios linfáticos reveló hiperplasia paracortical, necrosis en parches con abundantes restos celulares y una profusión de células histiocíticas periféricas. Se diagnosticó enfermedad de Kikuchi-Fujimoto (EKF) con compromiso cutáneo.
La EKF, también conocida como linfadenitis necrosante histiocítica, fue descrita, por primera vez por Kikuchi1 y Fujimoto2 en 1972. Se trata de un trastorno benigno y autolimitante caracterizado por linfadenopatía acompañada de fiebre baja y síntomas similares a los de la gripe. Suele haber compromiso de ganglios cervicales unilaterales y posteriores, si bien puede comenzar como una linfadenopatía generalizada3.
En mujeres la proporción es de más de 4:1. La predominancia de casos descritos en Japón y el hecho de que se hayan descrito, también, casos en muchas pacientes europeas y norteamericanas de ascendencia asiática, quizá apunte a cierta susceptibilidad racial o genética4. Su etiología no termina de estar clara.
Se sospecha, desde hace tiempo, de un origen vírico; no obstante, el curso clínico de esta enfermedad, la desaparición de lesiones sin tratamiento específico y cierta similitud con características propias del lupus eritematoso sistémico (LES) sugieren un mecanismo autoinmune subyacente3.
El diagnóstico se confirma a través de la biopsia de los ganglios linfáticos5. El compromiso de los ganglios linfáticos demuestra, característicamente, una arquitectura parcialmente obliterada por focos necróticos paracorticales confluentes, con abundantes restos cariorrécticos rodeados de histiocitos CD68+ e histiocitos MPO+, inmunoblastos, células T CD8+ y células dendríticas plasmacitoides CD123+6. Característicamente, no hay presencia de neutrófilos ni eosinófilos. La EKF se clasifica en tres subtipos histológicos distintos y, se cree, que avanza desde el tipo proliferativo (paracorteza expandida con aumento de histiocitos y células dendríticas plasmacitoides y restos nucleares cariorrécticos) hasta el tipo necrosante (predominancia de necrosis) para, finalmente, resolverse en el tipo xantomatoso (predominancia de histiocitos espumosos)6.
La piel es el órgano extranodal más comúnmente afectado, ya que se ha descrito compromiso cutáneo en el 16-40% de los pacientes7. Aunque algunos casos de lesiones cutáneas por EKF recuerdan a la urticaria, al sarpullido morbiliforme, a la rubéola, o a las erupciones por fármacos, la EKF cutánea suele comenzar con pápulas eritematosas y placas, predominantemente en rostro, brazos y parte superior del cuello. Los hallazgos histopatológicos de las biopsias cutáneas pueden recordar a los del lupus eritematoso discoide; no obstante, los restos cariorrécticos sin neutrófilos y la presencia de células CD68 y MPO positivas son característicos de la EKF7. Las lesiones cutáneas de la EKF tienden a resolverse en semanas o meses, así como la linfadenopatía.
Debido a la similitud de las características clínicas e histopatológicas de la EKF, el LES y el linfoma, no es inusual que los pacientes sean inicialmente mal diagnosticados. Por este motivo, identificar
los histiocitos característicos y los abundantes restos cariorrécticos y no las células del linfoma maligno o los cuerpos de hematoxilina patognomónicos de LES podría ser la clave del diagnóstico.
Sin embargo, está descrito que los sujetos con EKF son más susceptibles a desarrollar LES8; en concreto, la EKF puede preceder, suceder o coincidir con el diagnóstico de LES9. Aunque la relación que existe entre el LES y la EKF sigue siendo objeto de debate, se recomienda seguimiento periódico de los pacientes diagnosticados de EFK, para descartar la evolución a LES a lo largo de los años8.
La EKF suele ser autolimitante y se resuelve al cabo de 1-4 meses, aunque se ha descrito un índice de recurrencia de entre el 3% y el 4%. No existe un tratamiento específico para la EKF, si bien se utilizan analgésicos, antipiréticos y fármacos antiinflamatorios no esteroideos para aliviar la sensibilidad de los ganglios linfáticos y mitigar la fiebre. El uso de corticosteroides está recomendado para el tratamiento de cuadros de EKF generalizada o extranodal grave10, y si persisten los síntomas, se prescriben inmunoglobulinas intravenosas.
Nuestra paciente presentaba leucopenia y anticuerpos antinucleares positivos, si bien no cumplía los criterios establecidos por el Colegio Americano de Reumatología para ser diagnosticada de LES. Se le administró ibuprofeno oral durante tres meses y las linfadenopatías fueron desapareciendo, paulatinamente, así como la placa del cuero cabelludo. Tras un año de seguimiento, la tomografía computarizada solo reveló presencia de algunos ganglios linfáticos no específicos de pequeño tamaño.
La EKF es una enfermedad rara, sin embargo, es importante conocer esta entidad, ya que el correcto diagnóstico de la misma evitará realizar pruebas innecesarias, y tranquilizar a pacientes y a familiares dado su carácter autolimitado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


