
Inflammatory diseases of the muscles and skin are rare and orphan conditions. Dermatomyositis is an idiopathic inflammatory disorder associated with typical muscle and/or skin manifestations. Presentation with edema, ie, edematous dermatomyositis, is an infrequent variant. We report a new case of edematous dermatomyositis and review the literature.
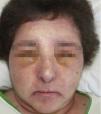
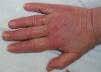
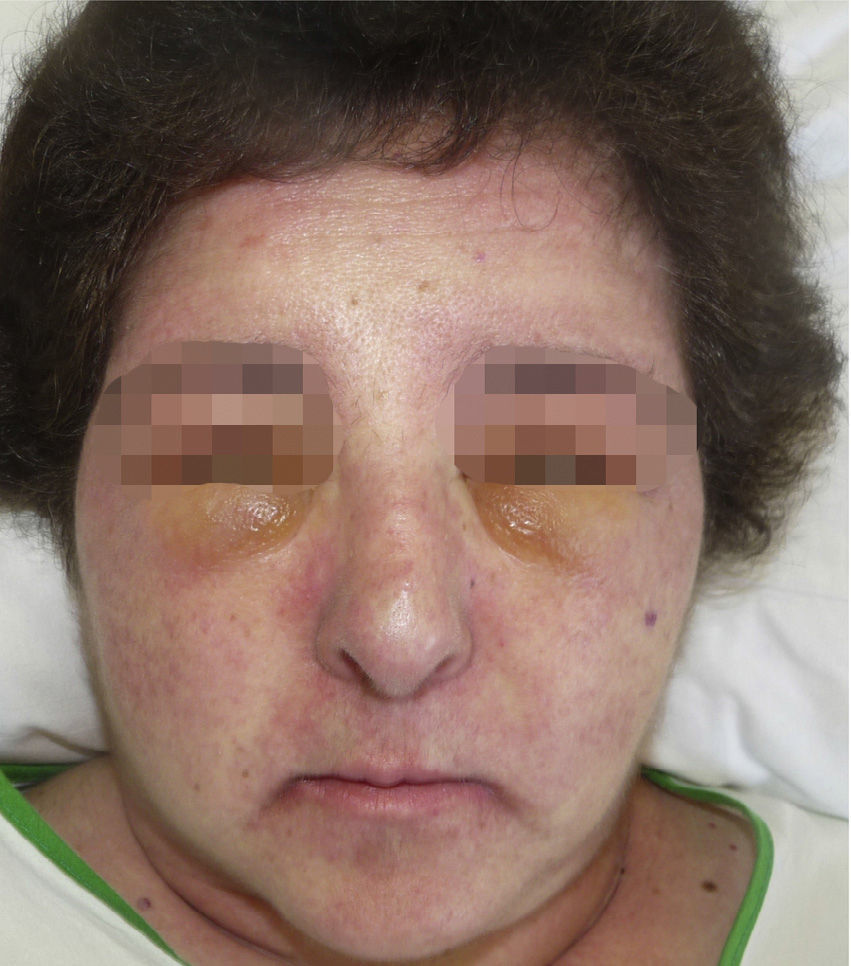
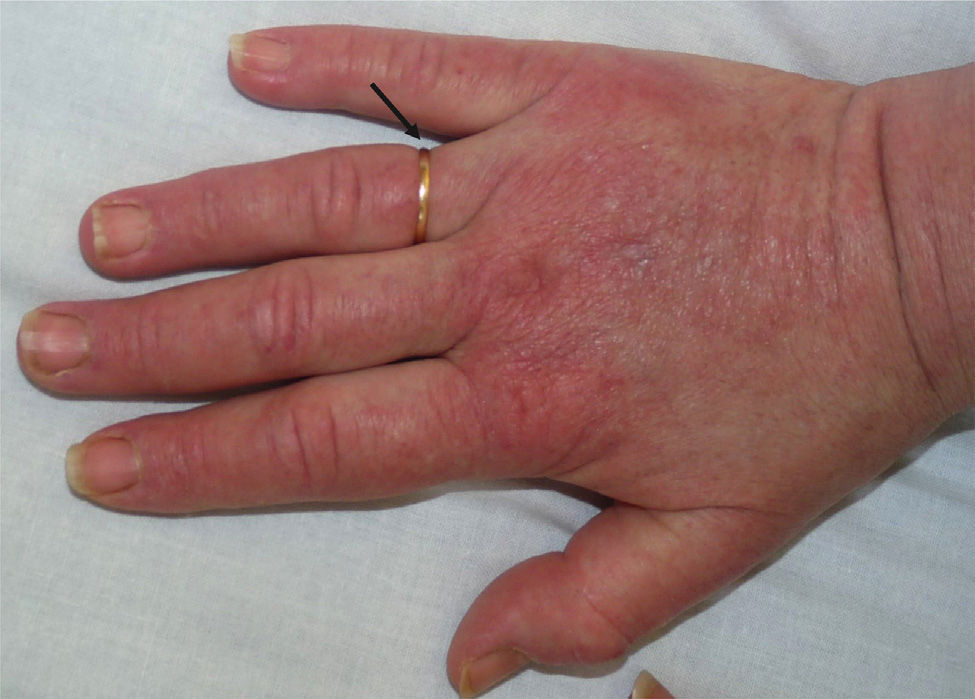
A 52-year-old woman with no clinical history of interest consulted with a pruriginous rash that first appeared 1 month previously. The rash took the form of violaceous erythema on the center of the face and around the eyes, erythematous macules on the dorsum of the metacarpophalangeal joints, and flagellate erythema on the back. The patient also experienced disabling muscle weakness that mainly affected proximal areas and intense dysphagia and dysphonia. The only findings of note in the laboratory workup were increased muscle and liver enzyme values, as follows: creatine phosphokinase, 4005IU/L (>140); aldolase, 44.4IU/L (>7.5); aspartate aminotransferase, 336IU/L (>31); alanine aminotransferase, 187IU/L (>40); γ-glutamyl transferase, 109IU/L (>30); and lactate dehydrogenase, 1022IU/L (>385). The results of antibody testing (antinuclear antibodies, anti-RNP, anti-Jo1, and anti-p155) were negative. Analysis of a skin biopsy specimen revealed vacuolar changes at the dermal-epidermal junction, solitary necrotic keratinocytes, and mucin deposits in the dermis. The electromyogram revealed signs of inflammatory myopathy, and occult underlying neoplasm was ruled out by tumor markers and positron emission tomography and computed tomography imaging. These findings confirmed the diagnosis of dermatomyositis, and treatment was started with intravenous methylprednisolone (1mg/kg/d); after 5 days, 4 doses of immunoglobulin (1.5g/kg/dose) were added, although there was little improvement. During the following months, the patient's condition progressed with intense edema affecting the face, neck, and upper extremities (visible on the magnetic resonance image) and myositis (Figs. 1–3). Therefore, methotrexate was added to the treatment regimen at 2 months, hydroxychloroquine at 4 months, and, given the lack of improvement, rituximab (1g) at 6 months in 2 doses separated by 2 weeks. The response was good, mainly in the skin. During follow-up, occasional thrombocytopenia and anemia (hemoglobin, 10.8g/dL; platelets, 58 000/μL) were recorded, as were increased values for indirect bilirubin (1mg/dL [>0.7]) and lactate dehydrogenase (641IU/L [>385]). These findings were compatible with Evans syndrome with dermatomyositis occurring alongside worsening muscle enzyme values. However, as the patient was receiving treatment with corticosteroids, it was impossible to perform the Coombs test to confirm this associated autoimmune etiology. Similarly, it was not possible to rule out other etiologies.
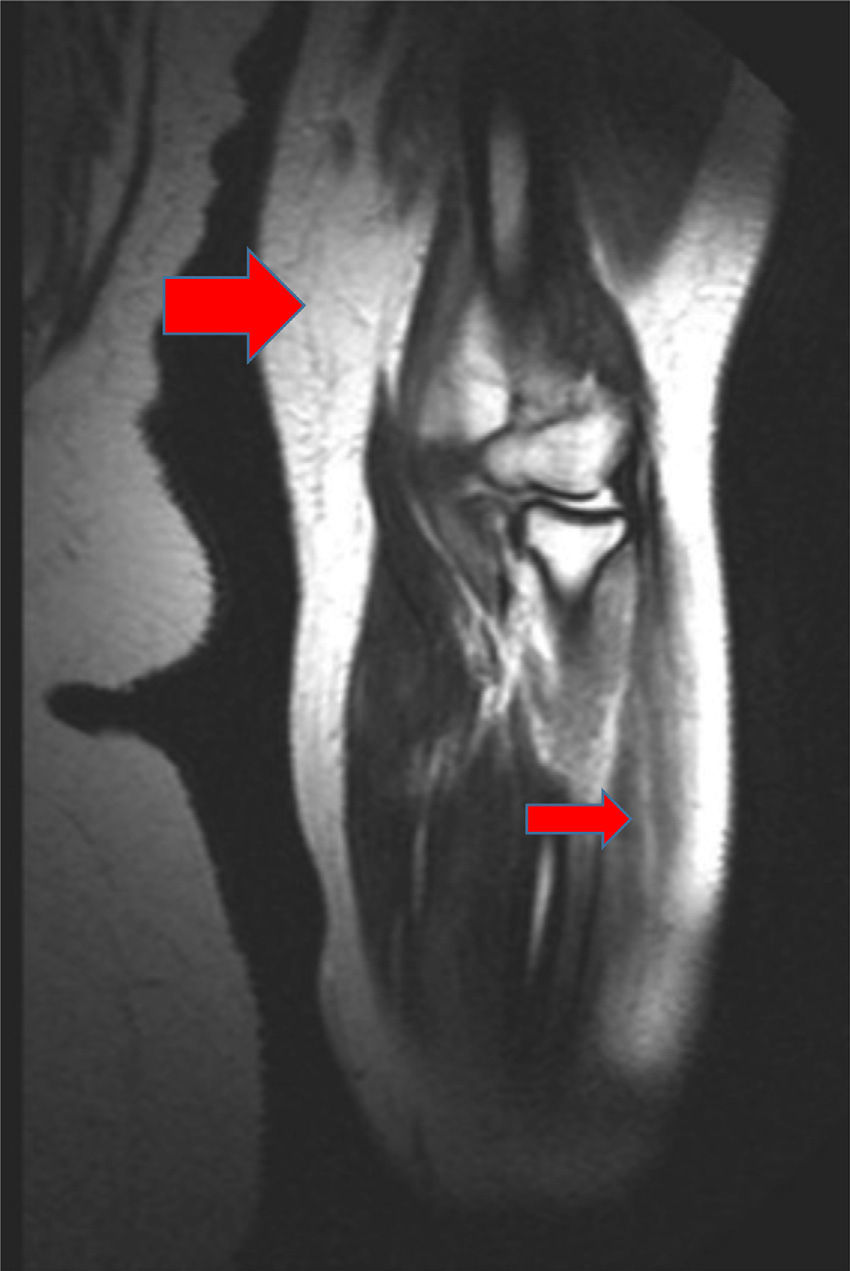
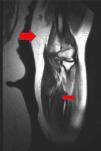
Magnetic resonance image of the left arm, where a hyperintense signal in several muscle groups is compatible with myositis. It is also possible to observe edema affecting the muscle and subcutaneous cellular tissue (the large red arrow points to an affected muscle group, and the small red arrow points to an area of edema in the subcutaneous cellular tissue).
Dermatomyositis is an autoimmune disease that mainly affects the skin and muscle. It has traditionally taken the form of mild periorbital edema accompanied by heliotrope rash. However, edematous dermatomyositis involves more extensive swelling. It is also a rare clinical variant of the condition, with only 23 cases reported in the literature.1 The etiology and pathogenesis remain unclear, although some authors have pointed to intense inflammatory activity with activation and deposition of complement, leading to vascular disease with muscle microinfarcts that in turn increase vascular permeability.1–5 This manifests clinically as edema affecting muscle and/or subcutaneous tissue, with or without pitting. Age at presentation is variable,1,2,6 with most adult cases occurring in women.1 Edema mainly affects the upper extremities, although it may be generalized4,6,7; cases of local edema have also been reported.2,8,9 Edematous dermatomyositis usually progresses more rapidly than classic dermatomyositis.5 Edema usually develops after skin involvement, although it may also be the initial presentation6; there have even been reports of edematous dermatomyositis with no other cutaneous findings.5 Muscle involvement and dysphagia are frequently associated with edematous dermatomyositis,1,4 as observed in the case we report.
It remains unclear whether this clinical presentation of dermatomyositis implies a greater risk of neoplasm5; according to a recently published literature review, the risk of neoplasm was recorded in fewer than 30% of cases (6 of 23 cases reviewed).1
Before a diagnosis of edematous dermatomyositis can be confirmed, it is important to rule out other, secondary causes of edema,1 such as kidney, heart, and thyroid disease, as well as hypoproteinemia. Treatment of edematous dermatomyositis should be intensive and early, given the potentially severe nature of the symptoms1,4,5,9,10 and the probable poorer prognosis than with classic dermatomyositis.4,6,10 The combination of high doses of intravenous corticosteroids and immunosuppressants seems to be a good alternative; immunoglobulins can be added when there is no response,3,5,8 and rituximab can be administered in refractory cases.1,2
Evans syndrome (anemia and/or autoimmune thrombocytopenia) rarely occurs with dermatomyositis. The first case was reported in 1990 in a woman with generalized edematous dermatomyositis.3 Although we were unable to confirm an autoimmune etiology in the case we report, the clinical and laboratory data were consistent with this diagnosis.
We report a new case of severe edematous dermatomyositis that proved refractory to several systemic treatments but responded well to rituximab. The condition probably occurred with Evans syndrome.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Flores-Terry MÁ, García-Arpa M, Anino-Fernández J, Mínguez-Sánchez MD. Dermatomiositis edematosa asociada a probable síndrome de Evans. Actas Dermosifiliogr. 2017;108:673–675.


