








la dermatomiositis (DM) se engloba dentro de las miopatías inflamatorias idiopáticas. La piel y el músculo esquelético son los órganos principalmente afectados. Un porcentaje significativo de pacientes, estimado entre un 15-25%, presentan un proceso neoplásico subyacente, aunque existen también formas exclusivamente cutáneas. Aún no se han identificado con exactitud qué factores predicen la evolución y el pronóstico de estos pacientes. En este trabajo aportamos muestra experiencia a partir de la descripción y análisis de una serie de pacientes.
Material y métodosestudio retrospectivo de 20 pacientes con diagnóstico de DM en seguimiento en el Servicio de Dermatología del Hospital General Universitario Gregorio Marañón, durante el periodo comprendido entre febrero de 2007 y febrero de 2010. Se evaluaron las características clínicas, histopatológicas, analíticas, pruebas complementarias y tratamientos realizados en dichos pacientes.
Resultadosdel total de la serie de pacientes 19 fueron mujeres. La edad media fue de 61 años (mediana 60). Identificamos 11 DM clásicas, 3 DM amiopáticas, 2 DM paraneoplásicas, 2 DM asociadas a enfermedad del tejido conectivo, una DM por fármacos y una DM juvenil. El eritema en heliotropo, las pápulas de Gottron y el eritema periungueal fueron las lesiones cutáneas más frecuentes. La necrosis cutánea estuvo presente en las dos pacientes con DM paraneoplásica. Los anticuerpos específicos de miositis fueron negativos en todos los pacientes. El tratamiento inicial fueron los corticoides sistémicos en el 85%. El 80% precisó la asociación de dos o más fármacos para el control de la enfermedad.
ConclusionesLa DM es un proceso potencialmente grave. El dermatólogo puede facilitar el diagnóstico y contribuir a detectar neoplasias asociadas y complicaciones sistémicas precozmente. La mayoría de los pacientes presentan un buen pronóstico, aunque requieren periodos de tratamiento prolongados. Los casos con más complicaciones son aquellos asociados a neoplasias y cuando existe compromiso cardiopulmonar.
Dermatomyositis is an idiopathic inflammatory myopathy that mainly affects the skin and skeletal muscle. An estimated 15% to 25% of patients have underlying tumors and some forms are exclusively cutaneous. The factors that predict disease course and prognosis in these patients have not been clearly identified. Here we report our experience through the description and analysis of a series of patients.
Material and methodsThis was a retrospective study of 20 patients with a diagnosis of dermatomyositis undergoing follow-up in the Department of Dermatology at Hospital General Universitario Gregorio Marañón in Madrid, Spain between February 2007 and February 2010. Clinical and histopathological characteristics were assessed alongside the results of laboratory tests and the treatments used.
ResultsNineteen of the 20 patients included in the study were women. The mean age was 61years (median, 60years). We identified 11 patients with classic, 3 with amyopathic, 2 with paraneoplastic, 1 with drug-associated, and 1 with juvenile dermatomyositis, and 2 patients had dermatomyositis associated with connective tissue disease. Heliotrope erythema, Gottron papules, and periungual erythema were the most frequent skin lesions. Cutaneous necrosis was present in 2 patients with paraneoplastic dermatomyositis. None of the patients had myositis-specific antibodies. Initial treatment was with systemic corticosteroids in 85% of cases. Eighty percent of patients required 2 or more drugs to achieve disease control.
ConclusionsDermatomyositis is a potentially serious disease. Dermatologists can facilitate diagnosis and contribute to the early detection of associated tumors and systemic complications. In most patients, the disease has a good prognosis, although extended periods of treatment may be required. Complications occur most commonly in patients with associated tumors or cardiopulmonary disease.
La dermatomiositis (DM) es una entidad infrecuente que se incluye dentro de las miopatías inflamatorias idiopáticas1. En 1975 Bohan y Peter establecieron una primera clasificación de las DM2. Posteriormente se han utilizado clasificaciones más amplias que incluyen nuevas entidades (tabla 1)1. Aún no existe unanimidad en cuanto al diagnóstico, las pruebas complementarias, el seguimiento y el tratamiento que debe realizarse en estos pacientes. Para el diagnóstico se emplean de forma habitual los criterios de Bohan y Peter, que incluyen debilidad muscular proximal, elevación sérica de las enzimas musculares, electromiograma con un patrón miopático, biopsia muscular patológica y lesiones cutáneas características2,3. En los últimos años la tendencia pasa por establecer una clasificación basada en la presencia de determinados anticuerpos. El hallazgo de algunos anticuerpos parece definir grupos homogéneos de pacientes que presentan características epidemiológicas, clínicas y un pronóstico similar4–6. Recientemente se ha demostrado que los pacientes con DM asociada a cáncer presentan los anticuerpos anti p-155 y anti p-155/140 con una mayor frecuencia que otras formas de miopatías inflamatorias7. Otros estudios tratan de correlacionar el tipo de lesiones cutáneas con diferentes formas clínicas de DM y con factores pronósticos8,9. Describimos nuestra experiencia en los pacientes con DM en seguimiento en nuestro departamento y discutimos sobre los algoritmos diagnósticos y terapéuticos.
Clasificación de las miopatías inflamatorias idiopáticas
| Dermatomiositis (DM) |
| Adulto |
| DM clásica* |
| DM clásica |
| DM clásica asociada a malignidad |
| DM clásica asociada a enfermedad del tejido conectivo |
| DM clínicamente amiopática*0,0**0,0*** |
| DM amiopática |
| DM hipomiopática |
| Juvenil |
| DM clásica |
| DM clínicamente amiopática |
| DM amiopática |
| DM hipomiopática |
| Polimiositis (PM) |
| PM |
| PM asociada a malignidad |
| PM asociada a enfermedad del tejido conectivo |
| Miositis por cuerpos de inclusión |
| Miositis por fármacos |
Adaptada de Sontheimer R1.
El término DM clásica se refiere a la presencia de clínica cutánea y muscular típicas. *La DM amiopática hace referencia a aquellos pacientes que presentan exclusivamente lesiones cutáneas en ausencia de enfermedad muscular; **la DM amiopática se considera provisional cuando no existe evidencia de afectación muscular en un periodo de al menos 6 meses. Se considera definitiva si este periodo es superior a 24 meses; ***los criterios de exclusión incluyen la ausencia de tratamiento con immunosupresores u otros fármacos conocidos capaces de producir lesiones cutáneas similares a DM.
Hemos realizado un estudio retrospectivo de los pacientes con diagnóstico de DM en seguimiento en el Servicio de Dermatología del Hospital General Universitario Gregorio Marañón, en el periodo comprendido entre febrero de 2007 y febrero de 2010. El diagnóstico de DM fue basado en los criterios de Bohan y Peter. Sólo los pacientes con diagnóstico definitivo o probable fueron incluidos. Los criterios de Sontheimer fueron utilizados para el diagnóstico de las formas amiopáticas1. La clasificación clínica utilizada se muestra en la tabla 1. Se incluyeron tanto los pacientes con nuevo diagnóstico como los que se encontraban en seguimiento en el momento del estudio.
ResultadosEn la serie presentada 19 pacientes eran mujeres y uno de ellos varón. La edad media fue de 61 años (mediana 60, rango 4-87). Once pacientes presentaron una DM clásica, tres una DM amiopática, dos una DM asociada a cáncer, dos una DM asociada a enfermedad del tejido conectivo, uno DM juvenil y uno DM asociada a fármacos (hidroxiurea). En la tabla 2 se muestran las características de cada uno de los pacientes. En 8 pacientes las lesiones cutáneas fueron la manifestación inicial de la enfermedad, en 5 pacientes fue la clínica muscular, en tres la clínica cutánea y muscular se manifestaron simultáneamente y 4 pacientes sólo presentaron clínica cutánea. El periodo de seguimiento fue de 32 meses de media (rango: 3-144 meses).
Características de los pacientes con dermatomiositis
| Sexo | Diagnóstico | Edad | Afectación sistémica | Anticuerpos | AP piel compatible | EMG | T° realizados | Otras características | |
| 1 | F | DM clásica | 32 | Mialgias y debilidad muscular | ANA | + | NR | CS, MTX | |
| 2 | F | DM clásica | 56 | Mialgias, febrícula, disfagia y disfonía | - | + | + | CS, MTX | |
| 3 | F | DM clásica | 26 | Mialgias, astenia | - | + | + | CS, MTX | Paniculitis lobulillar |
| 4 | F | DM clásica | 55 | Mialgias, astenia | ANA | + | + | CS, AZT, HCL | Ca 19.9 elevado |
| 5 | F | DM clásica | 53 | Mialgias | - | + | + | CS, MTX, HCL | |
| 6 | F | DM clásica | 58 | Mialgias, astenia, artralgias | - | + | + | CS. MTX, AZT | Paniculitis septal |
| 7 | F | DM clásica | 80 | Disfonía, disfagia, mialgias | - | + | + | CS, MTX, cloroquina | Carcinoma epidermoide parótida. Paniculitis lobulillar |
| 8 | F | DM clásica | 77 | Mialgias, artralgias | - | + | + | CS, MTX | TC con patrón en vidrio deslustrado. β-2 microglobulina↑ |
| 9 | F | DM clásica | 47 | Mialgias, artralgias | ANA | + | - | CS, MTX, AZR, HCL | |
| 10 | F | DM clásica | 76 | Mialgias, astenia | - | + | + | CS, MTX, AZT | |
| 11 | M | DM clásica | 62 | Mialgias, disfagia y disfonía | - | + | + | CS, MTX, talidomida | Carcinoma vesical in situ |
| 12 | F | DM amiopática | 87 | No | - | + | - | CS | Ca19.9 elevado |
| 13 | F | DM amiopática | 87 | No | ANA, Anti-PM-Scl | + | - | CS, MTX | |
| 14 | F | DM amiopática | 60 | No | - | + | - | CT | |
| 15 | F | DM asociada a ET | 60 | Artralgias, febrícula, EPID aguda | Anti-SSa/Ro | - | + | CS | Fallecimiento en 4 meses por EPID |
| 16 | F | DM asociada a ET | 83 | Mialgias, artralgias, astenia | ANA, Anti-U1RNP | + | + | CS, MTX, HCL, talidomida | Paniculitis lobulillar, alopecia en cuero cabelludo |
| 17 | F | DM paraneoplásica | 85 | Mialgias, astenia | - | + | + | CS | Adenocarcinoma endometrio |
| 18 | F | DM paraneoplásica | 54 | Mialgias, síndrome constitucional | ANA | + | + | CS, HCL | Carcinoma microcítico pulmón |
| 19 | F | DM juvenil | 4 | Mialgias, artralgias, fiebre | - | + | + | CS, MTX | Calcinosis |
| 20 | F | DM por fármacos | 80 | No | - | + | NR | CS | Sólo lesiones cutáneas |
AP: anatomía patológica; AZT: azatioprina; CS: ciclosporina; EMG: electromiograma; EPID: enfermedad pulmonar intersticial difusa; ET: enfermedad del tejido conectivo; HCL: hidroxicloroquina; NR: no realizado; MTX: metotrexato.
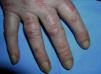
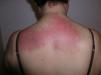
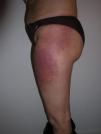
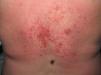
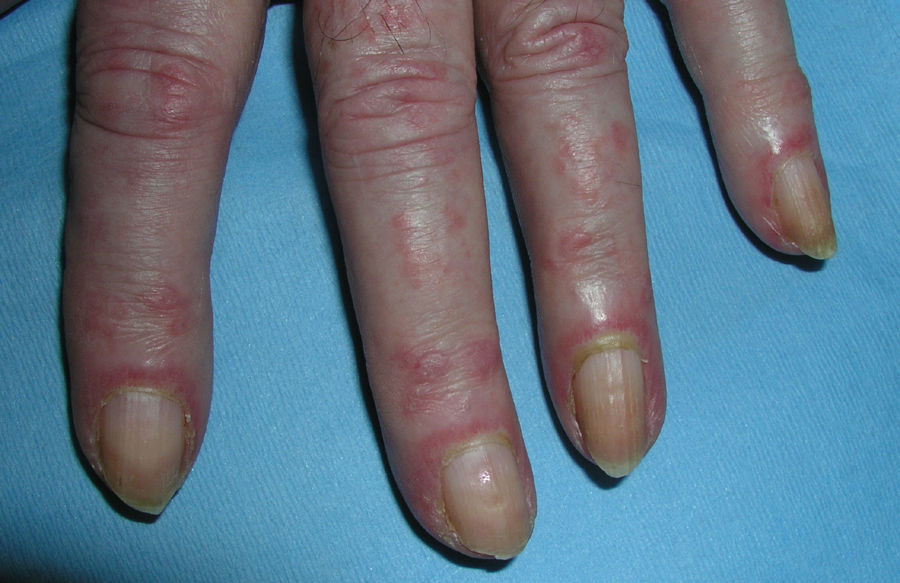
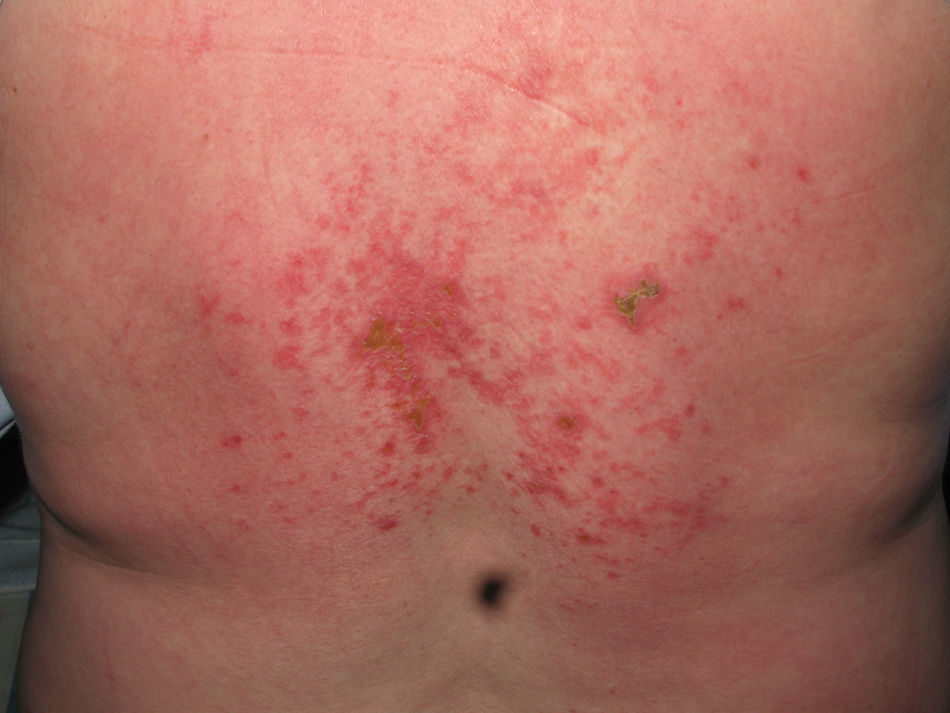
El eritema en heliotropo (fig. 1) y las pápulas de Gottron (fig. 2) estuvieron presentes en 14 y 12 de los pacientes, respectivamente. El hallazgo de un eritema violáceo macular confluente característico de la enfermedad fue frecuente en nuestra serie. El signo de la V torácica fue hallado en 11 pacientes. El signo del chal estuvo presente en 5 pacientes (fig. 3) y el signo de las pistoleras, cuando se afectan la cara externa de los muslos, en 5 (fig. 4). Otras áreas con un eritema violáceo macular confluente fueron la frente y la región malar en 8 pacientes y la cara externa de los brazos y los codos en 10. En las manos el área periungueal mostraba lesiones en la mayoría de los pacientes. El eritema periungueal estaba presente en 13 casos, las cutículas hipertróficas en 9 (fig. 5), las lesiones de calcinosis en dos y la necrosis en los pulpejos de los dedos en uno. El cuero cabelludo estaba afectado en tres pacientes, en dos en forma de alopecia difusa no cicatricial y en uno en forma de dermatitis psoriasiforme. Se detectaron lesiones cutáneas necróticas en el tronco en tres pacientes (fig. 6). Cuatro pacientes desarrollaron lesiones de paniculitis, que histológicamente se correspondían con una paniculitis lobulillar en tres casos y con una septal en otro. Cinco pacientes refirieron un empeoramiento de las lesiones cutáneas tras la exposición solar (tabla 3).
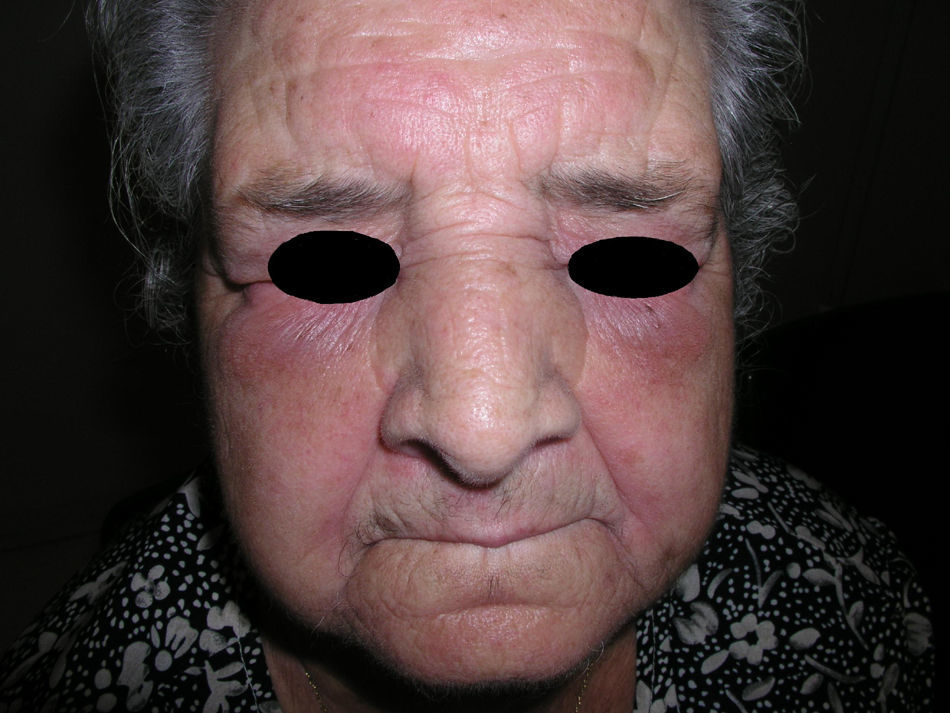
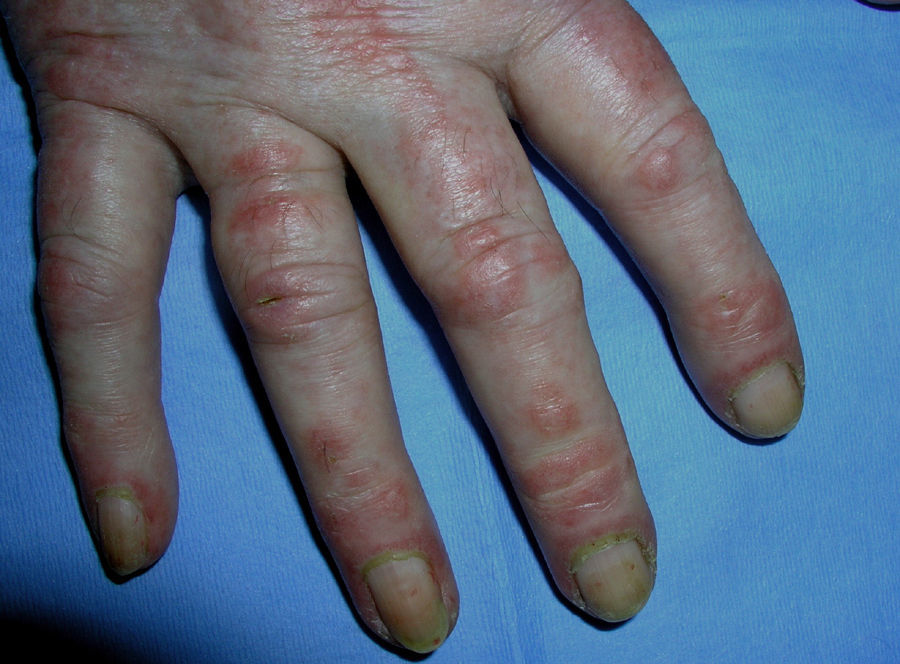
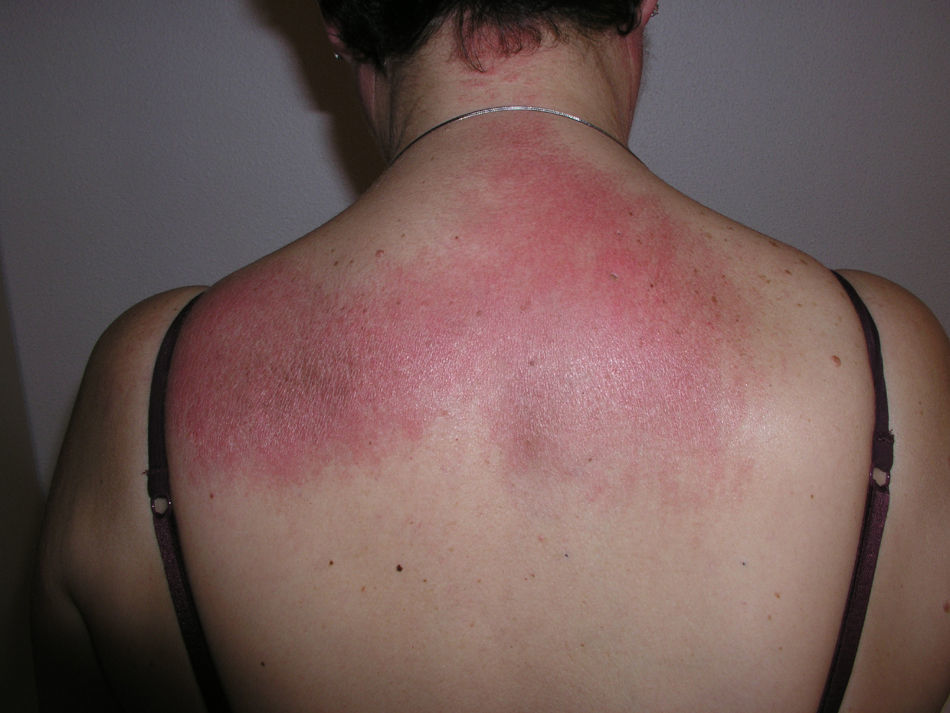
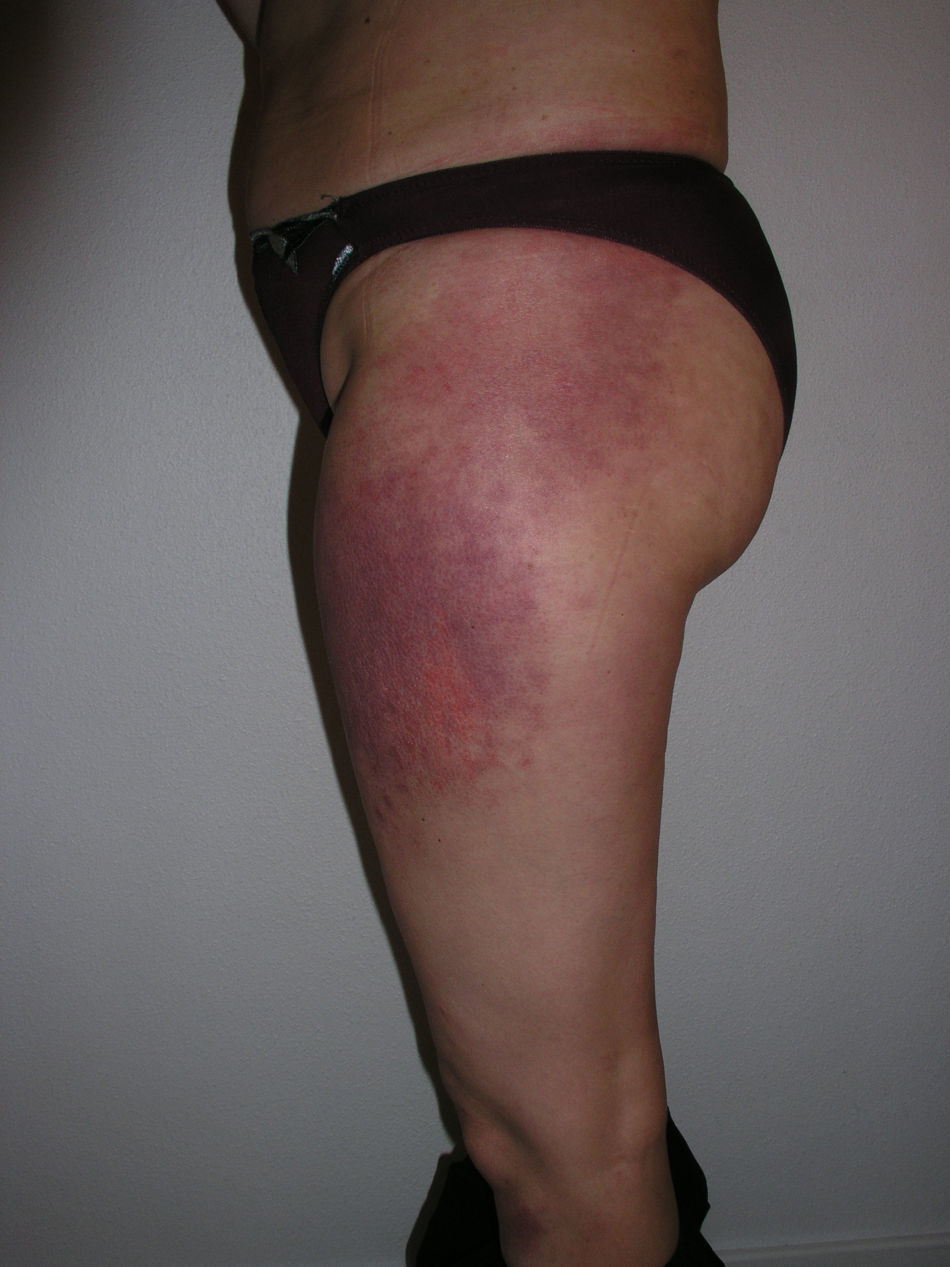
Lesiones cutáneas en los pacientes con dermatomiositis
| Lesiones cutáneas | Número de pacientes |
| Cabeza y cuello | |
| Eritema en heliotropo | 1,2,5,6,7,8,9,10,11,12,13,14,17,18 |
| Eritema violáceo en región frontal y malar | 7,11,15,17,18 |
| Dermatitis psoriasiforme en cuero cabelludo | 5 |
| Alopecia difusa no cicatricial | 11,16 |
| Tronco y extremidades | |
| Signo de la V torácica | 1,2,5,6,9,10,11,12,17,18,20 |
| Signo del chal | 5,6,9,10,20 |
| Signo de las pistoleras | 3,5,9,10,13 |
| Eritema violáceo en la cara externa de los brazos y en los codos | 2,3,5,7,9,13,16,17,18,20 |
| Lesiones necróticas | 5,17,18 |
| Manos | |
| Pápulas de Gottron | 1,2,4,5,6,7,8,9,10,11,17,18 |
| Eritema periungueal | 1,4,6,7,9,11,12,13,14,15,17,18,19 |
| Cutículas hipertróficas | 4,7,9,11,12,14,17,18,19 |
| Calcinosis | 15,19 |
| Necrosis en los pulpejos de los dedos | 16 |
| Otras | |
| Paniculitis | 3,6,7,16 |
| Manos de mecánico | |
| Poiquilodermia | |
Quince pacientes presentaron mialgias generalizadas y debilidad muscular proximal, 6 tuvieron artralgias, tres manifestaron disfagia y disfonía y dos desarrollaron una enfermedad pulmonar intersticial difusa (EPID). Una de las pacientes con EPID falleció en 4 meses por la enfermedad pulmonar.
Exploraciones complementariasLas enzimas de daño muscular (creatina quinasa, aldolasa, lactato deshidrogenasa, aspartato aminotransferasa y alanina aminotransferasa) se detectaron elevadas en 15 pacientes. Los anticuerpos anti nucleares (ANA) fueron positivos en 6 de los casos. Los anticuerpos específicos de miositis, incluyendo los anticuerpos anti-Jo1, anti-PL-12, anti-PL-7, anti-Mi-2 y anti-PRS fueron negativos en todos los pacientes. De los anticuerpos asociados a miositis los anti-PM-Scl, los anti-U1-RNP y los anti-SSA/RO fueron positivos en un paciente cada uno, siendo el resto negativos. Los marcadores de antígenos tumorales que se midieron incluían el CA19.9, CA125, PSA, CYFRA, CEA y β-2-microglobulina. En dos pacientes se detectó el CA19.9 elevado con valores de 50 y 55,7 (valor normal hasta 19 U/ml). En otra paciente se encontró una β-2-microglobulina de 4 (valor normal hasta 3,2mg/l). La biopsia cutánea, realizada en todos los casos, fue compatible con DM en 18 pacientes. En 10 pacientes la biopsia de una pápula de Gottron mostró una hiperqueratosis variable, con acantosis y degeneración vacuolar de la basal. En la dermis podía observarse ectasia vascular y un infiltrado inflamatorio perivascular. En 8 pacientes la biopsia de áreas de eritema violáceo mostró una dermatitis de interfase con degeneración vacuolar de la basal y atrofia epidérmica. En 11 muestras histológicas se encontraron depósitos de mucina en la dermis. El electromiograma, que fue realizado en 18 casos, mostró un patrón de afectación miopática en 14 pacientes. La biopsia muscular, que fue practicada en 12 casos, mostró un patrón de miositis en 9 pacientes.
TratamientoEl tratamiento inicial fueron los corticoides sistémicos en 17 pacientes a una dosis de 0,5-1mg/kg/día durante una media de 4 meses. Se continuó con dosis<0,5mg/kg/día durante una media de 5 meses en 10 pacientes. Los pacientes en tratamiento con corticoides sistémicos durante más de dos meses desarrollaron un hábito cushingoide. En 16 casos fue necesario asociar otros fármacos. Metotrexato fue utilizado en 14 pacientes a una dosis de 15mg/semana, con una media de duración de 12 meses. En un paciente tuvo que ser suspendido por intolerancia gástrica. La hidroxicloroquina se administró a una dosis de 400mg/día, durante una media de 14 meses en 5 pacientes. Otros tratamientos utilizados vienen reflejados en la tabla 4.
Tratamiento en los pacientes con dermatomiositis
| Tratamiento | Dosis | Duración | Mejoría | Efectos adversos | N° pacientes |
| Corticoides sistémicos | 0,5-1 mg/kg/d | 4 meses | Cutánea y muscular | Hábito cushingoide en 7 pacientes | 17 |
| < 0,5 mg/kg/d | 5 meses | 10 | |||
| Metotrexato | 15 mg/smn | 12 meses | Cutánea | Intolerancia gástrica en 1 paciente | 14 |
| Azatioprina | 100 mg/d | 6 meses | Cutánea y muscular | - | 4 |
| Hidroxicloroquina | 400 mg/d | 14 meses | Cutánea | - | 5 |
| Cloroquina | 250 mg/d | 3 meses | Cutánea | Visión borrosa en 1 paciente | 1 |
| Talidomida | 100 mg/d | 5 meses | Cutánea | - | 2 |
Durante el periodo de seguimiento 4 pacientes desarrollaron neoplasias malignas. Una paciente había presentado un adenocarcinoma de endometrio tres años antes del diagnóstico de la DM. Coincidiendo con una recidiva del proceso maligno, se inició la clínica de la DM. La paciente fallecía tres meses después por metástasis diseminadas. Otra paciente fue diagnosticada de un carcinoma microcítico de pulmón dos meses tras el inicio de la DM. Presentó buena respuesta al tratamiento oncológico, mejorando simultáneamente las lesiones cutáneas y la debilidad muscular. Tras 8 meses de remisión tumoral no existía evidencia de actividad cutánea ni muscular. Otra paciente desarrolló un carcinoma epidermoide de parótida 18 meses después del diagnóstico de la DM. Tras 20 meses de remisión tumoral completa continuaba con lesiones cutáneas características de DM y con debilidad muscular. Otro paciente fue diagnosticado de un carcinoma in situ de vejiga. A los 24 meses, a pesar de estar libre de enfermedad tumoral, persistían las lesiones cutáneas.
DiscusiónEn nuestra serie la DM clásica supuso la forma de presentación más frecuente, observada en el 55% del total. Las formas amiopática y la asociada a cáncer significaron un 15 y un 10%, respectivamente. Estos datos son similares a los descritos en otros estudios3,10,11. Se observó un predominio en el sexo femenino. Se ha documentado una relación mujer:hombre de 2:11. En nuestra serie fue superior, ya que tan sólo se incluye un paciente masculino. Sin embargo, el escaso número de pacientes de la serie impide extraer conclusiones en este punto.
Las lesiones cutáneas más frecuentes fueron el eritema en heliotropo y las pápulas de Gottron. Otras manifestaciones observadas en más de la mitad de los pacientes fueron el eritema violáceo macular confluyente y el eritema periungueal. El eritema violáceo en las zonas fotoexpuestas y en las áreas de extensión, junto con lesiones en el dorso de las manos y el área periungueal, debe hacer sospechar el diagnóstico de DM. Los signos más característicos y que permiten distinguir la DM del lupus eritematoso son la coloración violácea del eritema y la tendencia de las lesiones a distribuirse en torno a los ojos y en las zonas extensoras. La calcinosis cutánea se presentó en los dedos de las manos en dos pacientes. Una de ellas fue clasificada como una DM juvenil. Este hallazgo es más frecuente en pacientes jóvenes12. Tres pacientes desarrollaron lesiones necróticas en el tronco; de ellas, dos presentaron una DM asociada a cáncer. Este tipo de lesiones han sido descritas con mayor frecuencia en pacientes con DM paraneoplásica, por lo que su presencia debe aumentar la sospecha de que el paciente asocie una neoplasia maligna8,9.
Las artralgias fueron consideradas como leves en la mayoría de los casos. En dos pacientes se desarrolló una EPID; uno de ellos presentó una evolución aguda con disnea e hipoxemia severas y fallecimiento a los 4 meses. En el otro caso la TC mostraba un patrón en vidrio deslustrado, pero la paciente permaneció asintomática durante el periodo de seguimiento. Se ha descrito afectación pulmonar hasta en el 40% de los pacientes con DM. En muchas ocasiones estas alteraciones sólo son evidentes en las pruebas de imagen13–17. Otros tres pacientes presentaron disfagia y disfonía, que se resolvieron rápidamente tras los primeros meses de tratamiento. La afectación cardíaca se ha descrito hasta en el 50% de los pacientes con DM, pero sólo una pequeña proporción manifiesta síntomas18. La afectación gastrointestinal es más frecuente en las formas juveniles19. En nuestra serie ningún paciente presentó alteraciones a nivel de estos órganos.
Las enzimas de daño muscular se elevaron en la mayoría de los pacientes. Siguieron un patrón aleatorio y no se distinguió una tendencia en ningún parámetro. Algunos pacientes se encontraban ya en tratamiento con corticoides cuando se realizaron las primeras analíticas, por lo que algunos valores podrían estar enmascarados. Todos los anticuerpos específicos de miositis fueron negativos. Son considerados muy específicos de DM, pero su sensibilidad es baja, por lo que su ausencia no puede descartar el diagnóstico de esta enfermedad5,20,21. Respecto a los anticuerpos asociados a miositis, el anti-U1RNP y el anti-SSA/RO fueron positivos en dos pacientes con síndrome de solapamiento. El anti-PM-Scl fue positivo en una paciente con DM amiopática. Tres pacientes presentaron marcadores tumorales algo elevados. En ninguno de ellos, tras periodos de seguimiento prolongados, se detectó enfermedad tumoral.
La incidencia de cáncer en pacientes con DM oscila entre el 9 y el 42%7–9,22. Si se consideran los criterios de DM asociada a cáncer de Bohan y Peter (la neoplasia aparece alrededor de los tres años del diagnóstico inicial de la DM y desaparece una vez erradicado el tumor), tan sólo se pueden clasificar pacientes con DM paraneoplásicas a 2 de los 4 con tumores malignos6. La primera paciente inició la DM coincidiendo con una recidiva tumoral y falleció pocos meses después. En la segunda paciente los signos y síntomas de la DM prácticamente desaparecieron cuando finalizó el tratamiento oncológico. Estos dos casos sí siguieron un curso paraneoplásico. En cambio, en los dos últimos pacientes, a pesar del periodo prolongado de remisión tumoral, persistían las lesiones características de la DM. El diagnóstico temprano de la DM y la realización del estudio de extensión probablemente contribuyeran a un diagnóstico más precoz de las neoplasias. La definición de unos factores predictivos de malignidad permitiría seleccionar con mayor precisión a aquellos pacientes en los que el estudio exhaustivo de patología tumoral fuera más prioritario. Se han descrito factores de riesgo independientes para presentar una neoplasia subyacente; entre ellos se incluyen una edad>52 años en el momento del diagnóstico, un inicio abrupto y rápido de las lesiones cutáneas y de la debilidad muscular, la presencia de lesiones necróticas y de eritema periungueal y unos niveles bajos de C4. Otros parámetros encontrados con mayor frecuencia en estos pacientes son valores elevados de la enzima creatina quinasa (>1.000 UI/l) y títulos elevados de los anticuerpos p155/140. En cambio, se ha considerado como factor protector unas cifras bajas de linfocitos (< 1.500/mm3)8.
Actualmente no existe un consenso sobre qué tipo de exploraciones deben realizarse en los pacientes con DM para descartar una neoplasia asociada. Una opción razonable incluiría, en varones, un examen clínico completo, un análisis de laboratorio de rutina, unos marcadores de antígenos tumorales y una TAC de tórax y abdomen. En mujeres sería conveniente también realizar una mamografía y una ecografía pélvica. El estudio del tracto intestinal mediante endoscopia vendría indicado por la clínica y la edad del paciente5,23,24.
Los pacientes con DM asocian síntomas de otras colagenosis hasta en el 20% de los casos2. En la DM asociada a otra enfermedad del tejido conectivo las lesiones cutáneas y musculares de la DM se asocian a otros datos de colagenosis. Los dos pacientes que tuvieron esta forma de DM presentaron mayor afectación sistémica y múltiples lesiones cutáneas.
Asimismo, la DM puede tener un origen farmacológico, siendo la hidroxiurea, un fármaco utilizado en el tratamiento de enfermedades mieloproliferativas crónicas, una de las causas medicamentosas de erupción DM-like más conocidas. Estos pacientes presentan lesiones cutáneas sin afectación muscular ni sistémica. Como en el caso que describimos, la erupción cutánea puede aparecer tras muchos años de tratamiento25.
Los corticoides sistémicos continúan siendo el tratamiento de elección en los pacientes con DM11. Existe una clara discordancia entre la respuesta al tratamiento de la afectación muscular y la respuesta de la afectación cutánea. Los corticoides sistémicos se retiraron progresivamente según mejoraba la clínica muscular y sistémica. La clínica muscular mejoró de forma significativa a partir del primer mes de tratamiento. En cambio, las lesiones cutáneas persistieron durante más de 6 meses en más de la mitad de los pacientes. El tratamiento de las manifestaciones cutáneas continúa siendo poco satisfactorio. En los primeros meses de tratamiento la tendencia es la de introducir fármacos ahorradores de corticoides26,27. El más utilizado fue el metotrexato, seguido de la hidroxicloroquina. Las lesiones cutáneas presentaron buena respuesta a dichos tratamientos. En un estudio reciente se demostró que el uso temprano de metotrexato a una dosis de 15mg/semana permitía un descenso más rápido de los corticoides sistémicos y con menos efectos secundarios27. La azatioprina se utilizó en pacientes refractarios o con mala tolerancia al metotrexato con respuesta parcial. La talidomida fue utilizada en dos pacientes refractarios a los tratamientos previos con buena tolerancia y respuesta en las lesiones cutáneas. Sin embargo, aún no existe un protocolo validado para el tratamiento de la DM.
Los datos descritos en nuestra serie son similares a los de estudios previos, exceptuando el marcado predominio femenino. Para el diagnóstico las lesiones cutáneas características, los hallazgos de las biopsias cutánea y muscular y del electromiograma fueron datos concluyentes en la mayoría de los casos. El pronóstico fue favorable, excepto en aquellos casos asociados a enfermedad pulmonar grave y en los vinculados a una neoplasia maligna. La respuesta terapéutica fue buena en la mayoría de los casos, aunque se requirieron periodos prolongados de tratamiento. Los pacientes con DM paraneoplásica eran mayores de 50 años, iniciaron la clínica de la DM de forma rápida y presentaron lesiones cutáneas necróticas. En estos casos parece recomendable hacer más exhaustiva la búsqueda de una posible neoplasia asociada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


