







Dermatomyositis is an idiopathic inflammatory myopathy that mainly affects the skin and skeletal muscle. An estimated 15% to 25% of patients have underlying tumors and some forms are exclusively cutaneous. The factors that predict disease course and prognosis in these patients have not been clearly identified. Here we report our experience through the description and analysis of a series of patients.
Material and methodsThis was a retrospective study of 20 patients with a diagnosis of dermatomyositis undergoing follow-up in the Department of Dermatology at Hospital General Universitario Gregorio Marañón in Madrid, Spain between February 2007 and February 2010. Clinical and histopathological characteristics were assessed alongside the results of laboratory tests and the treatments used.
ResultsNineteen of the 20 patients included in the study were women. The mean age was 61years (median, 60years). We identified 11 patients with classic, 3 with amyopathic, 2 with paraneoplastic, 1 with drug-associated, and 1 with juvenile dermatomyositis, and 2 patients had dermatomyositis associated with connective tissue disease. Heliotrope erythema, Gottron papules, and periungual erythema were the most frequent skin lesions. Cutaneous necrosis was present in 2 patients with paraneoplastic dermatomyositis. None of the patients had myositis-specific antibodies. Initial treatment was with systemic corticosteroids in 85% of cases. Eighty percent of patients required 2 or more drugs to achieve disease control.
ConclusionsDermatomyositis is a potentially serious disease. Dermatologists can facilitate diagnosis and contribute to the early detection of associated tumors and systemic complications. In most patients, the disease has a good prognosis, although extended periods of treatment may be required. Complications occur most commonly in patients with associated tumors or cardiopulmonary disease.
la dermatomiositis (DM) se engloba dentro de las miopatías inflamatorias idiopáticas. La piel y el músculo esquelético son los órganos principalmente afectados. Un porcentaje significativo de pacientes, estimado entre un 15-25%, presentan un proceso neoplásico subyacente, aunque existen también formas exclusivamente cutáneas. Aún no se han identificado con exactitud qué factores predicen la evolución y el pronóstico de estos pacientes. En este trabajo aportamos muestra experiencia a partir de la descripción y análisis de una serie de pacientes.
Material y métodosestudio retrospectivo de 20 pacientes con diagnóstico de DM en seguimiento en el Servicio de Dermatología del Hospital General Universitario Gregorio Marañón, durante el periodo comprendido entre febrero de 2007 y febrero de 2010. Se evaluaron las características clínicas, histopatológicas, analíticas, pruebas complementarias y tratamientos realizados en dichos pacientes.
Resultadosdel total de la serie de pacientes 19 fueron mujeres. La edad media fue de 61 años (mediana 60). Identificamos 11 DM clásicas, 3 DM amiopáticas, 2 DM paraneoplásicas, 2 DM asociadas a enfermedad del tejido conectivo, una DM por fármacos y una DM juvenil. El eritema en heliotropo, las pápulas de Gottron y el eritema periungueal fueron las lesiones cutáneas más frecuentes. La necrosis cutánea estuvo presente en las dos pacientes con DM paraneoplásica. Los anticuerpos específicos de miositis fueron negativos en todos los pacientes. El tratamiento inicial fueron los corticoides sistémicos en el 85%. El 80% precisó la asociación de dos o más fármacos para el control de la enfermedad.
ConclusionesLa DM es un proceso potencialmente grave. El dermatólogo puede facilitar el diagnóstico y contribuir a detectar neoplasias asociadas y complicaciones sistémicas precozmente. La mayoría de los pacientes presentan un buen pronóstico, aunque requieren periodos de tratamiento prolongados. Los casos con más complicaciones son aquellos asociados a neoplasias y cuando existe compromiso cardiopulmonar.
Dermatomyositis is a rare idiopathic inflammatory myopathy.1 It was initially classified by Bohan and Peter in 1975.2 Broader classifications have since been used that include new entities (Table 1).1 Consensus regarding diagnosis, additional tests, follow-up, and treatment of these patients is not yet unanimous. Diagnosis is usually performed using the Bohan and Peter criteria, which include proximal muscle weakness, raised serum levels of muscle enzymes, electromyography showing a myopathic pattern, a muscle biopsy showing disease, and characteristic skin lesions.2,3 In recent years, there has been a tendency to establish classification based on the presence of specific antibodies. The presence of certain antibodies appears to define homogeneous groups of patients with similar epidemiologic and clinical characteristics and a similar prognosis.4–6 It has recently been shown that anti-p155 and anti-p155/140 antibodies are more commonly found in patients with dermatomyositis associated with cancer than in those with other forms of inflammatory myopathy.7 Other studies have attempted to correlate the type of skin lesion with different clinical forms of dermatomyositis and with prognostic factors.8,9 Here we describe a series of patients with dermatomyositis seen in our department and discuss the appropriateness of diagnostic and therapeutic algorithms for the disease.
Classification of Idiopathic Inflammatory Myopathy.
| Dermatomyositis | |
| Adult | |
| Classic DMa | |
| Classic DM | |
| Classic DM associated with cancer | |
| Classic DM associated with connective tissue disease | |
| Clinically amyopathic DMb,c,d | |
| Amyopathic DM | |
| Hypomyopathic DM | |
| Juvenile | |
| Classic DM | |
| Clinically amyopathic DM | |
| Amyopathic DM | |
| Hypomyopathic DM | |
| Polymyositis | |
| PM | |
| PM associated with cancer | |
| PM associated with connective tissue disease | |
| Inclusion body myositis | |
| Drug induced myositis |
Adapted from Sontheimer R.1.
We performed a retrospective study of patients diagnosed with dermatomyositis who were undergoing follow-up in the Department of Dermatology at Gregorio Marañón General University Hospital in Madrid, Spain between February 2007 and February 2010. Diagnosis of dermatomyositis was based on the Bohan and Peter criteria. Only patients with a definitive or probable diagnosis were included. The criteria described by Sontheimer1 were used to diagnose amyopathic dermatomyositis. The clinical classification used in the study is shown in Table 1. Patients with a new diagnosis and those who were in follow-up at the time of the study were included.
ResultsThe study included 19 women and 1 man. The mean age was 61 years (median, 60 y; range, 4-87 y). Eleven patients had classic dermatomyositis, 3 amyopathic dermatomyositis, 2 had dermatomyositis associated with cancer, 2 dermatomyositis associated with connective-tissue disease, 1 had juvenile dermatomyositis, and 1 had drug-induced dermatomyositis linked to the use of hydroxyurea. Table 2 shows the characteristics of each patient. The disease manifested initially as skin lesions in 8 patients and as muscle symptoms in 5 patients, whereas in 3 patients, muscle and skin symptoms manifested simultaneously. Four patients presented only skin symptoms. The mean follow-up time was 32 months (range, 3-144 months).
Characteristics of Patients with Dermatomyositis.
| Sex | Diagnosis | Age, y | Systemic Involvement | Antibodies | Compatible Skin HP | EMG | Treatments Performed | Other Characteristics | |
| 1 | F | Classic DM | 32 | Myalgia and muscle weakness | ANA | + | NP | CS, MTX | |
| 2 | F | Classic DM | 56 | Myalgia, low-grade fever, dysphagia, and dysphonia | - | + | + | CS, MTX | |
| 3 | F | Classic DM | 26 | Myalgia, asthenia | - | + | + | CS, MTX | Lobular panniculitis |
| 4 | F | Classic DM | 55 | Myalgia, asthenia | ANA | + | + | CS, AZT, HCL | Elevated CA19-9 |
| 5 | F | Classic DM | 53 | Myalgia | - | + | + | CS, MTX HCL | |
| 6 | F | Classic DM | 58 | Myalgia, asthenia, arthralgia | - | + | + | CS MTX, AZT | Septal panniculitis |
| 7 | F | Classic DM | 80 | Dysphonia, dysphagia, myalgia | - | + | + | CS, MTX, chloroquine | Parotid epidermoid carcinoma Lobular panniculitis |
| 8 | F | Classic DM | 77 | Myalgia, arthralgia | - | + | + | CS, MTX | CT with ground glass opacity, elevated β-2 microglobulin |
| 9 | F | Classic DM | 47 | Myalgia, arthralgia | ANA | + | - | CS, MTX, AZT, HCL | |
| 10 | F | Classic DM | 76 | Myalgia, asthenia | - | + | + | CS, MTX, AZT | |
| 11 | M | Classic DM | 62 | Myalgia, dysphagia, and dysphonia | - | + | + | CS, MTX, thalidomide | Carcinoma in situ of the bladder |
| 12 | F | Amyopathic DM | 87 | No | - | + | - | CS | Elevated CA19-9 |
| 13 | F | Amyopathic DM | 87 | No | ANA, Anti-PM-Scl | + | - | CS, MTX | |
| 14 | F | Amyopathic DM | 60 | No | - | + | - | CS | |
| 15 | F | DM associated with CTD | 60 | Arthralgia, low-grade fever, acute DILD | Anti-SSA/RO | - | + | CS | Death in 4 months due to DILD |
| 16 | F | DM associated with CTD | 83 | Myalgia, arthralgia, asthenia | ANA, Anti-U1RNP | + | + | CS, MTX, HCL, thalidomide | Lobular panniculitis, alopecia on the scalp |
| 17 | F | Paraneoplastic DM | 85 | Myalgia, asthenia | - | + | + | CS | Endometrial adenocarcinoma |
| 18 | F | Paraneoplastic DM | 54 | Myalgia, constitutional syndrome | ANA | + | + | CS, HCL | Small-cell lung carcinoma |
| 19 | F | Juvenile DM | 4 | Myalgia, arthralgia, fever | - | + | + | CS, MTX | Calcinosis |
| 20 | F | Drug induced DM | 80 | No | - | + | NP | CS | Skin lesions only |
Abbreviations: AZT, azathioprine; CS, ciclosporin; CTD, connective tissue disease; DILD, diffuse interstitial lung disease; DM, dermatomyositis; EMG, electromyography; HCL, hydroxychloroquine; HP, histopathology; MTX, methotrexate; NP, not performed.
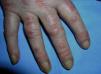
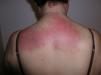
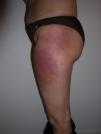
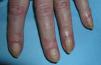
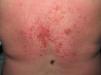
Heliotrope erythema (Fig. 1) and Gottron papules (Fig. 2) were found in 14 and 12 patients, respectively. Many of the patients had confluent macular violaceous erythema, which is characteristic of the disease. The V-sign on the chest was found in 11 patients. The shawl sign was found in 5 patients (Fig. 3) and the holster sign, on the outer surface of the thighs, in 5 patients (Fig. 4). Confluent macular violaceous erythema was also found on the forehead and cheeks in 8 patients and on the outer surface of the arms and elbows in 10 patients. Lesions of the periungual region were found on the hands of most patients. Periungual erythema was found in 13 patients, hypertrophic cuticles in 9 (Fig. 5), calcinosis lesions in 2, and necrosis of the soft parts of the fingers in 1. The scalp was affected in 3 patients: 2 in the form of diffuse nonscarring alopecia and 1 in the form of psoriasiform dermatitis. Necrotic skin lesions were found on the torso of 3 patients (Fig. 6). Four patients developed panniculitis lesions; histology revealed lobular panniculitis in 3 cases and septal panniculitis in 1 case. Five patients reported deterioration of skin lesions after exposure to sunlight (Table 3).
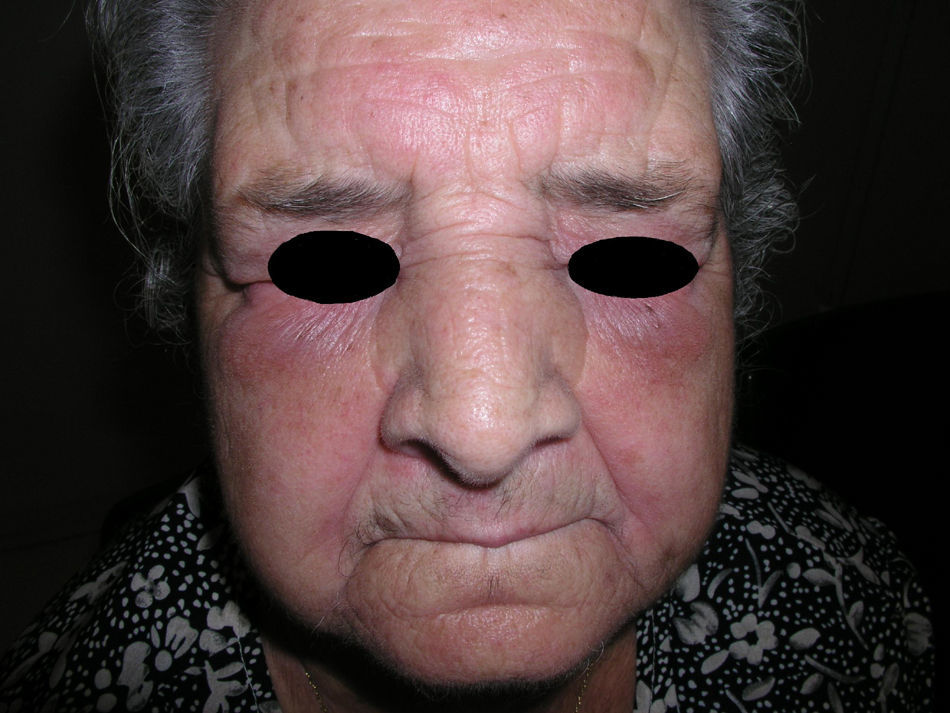
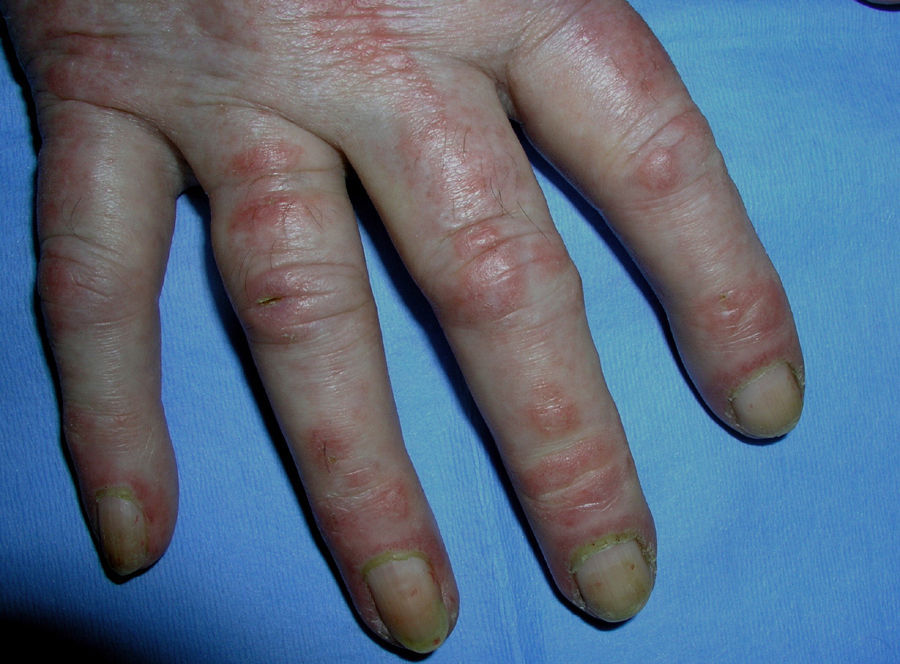
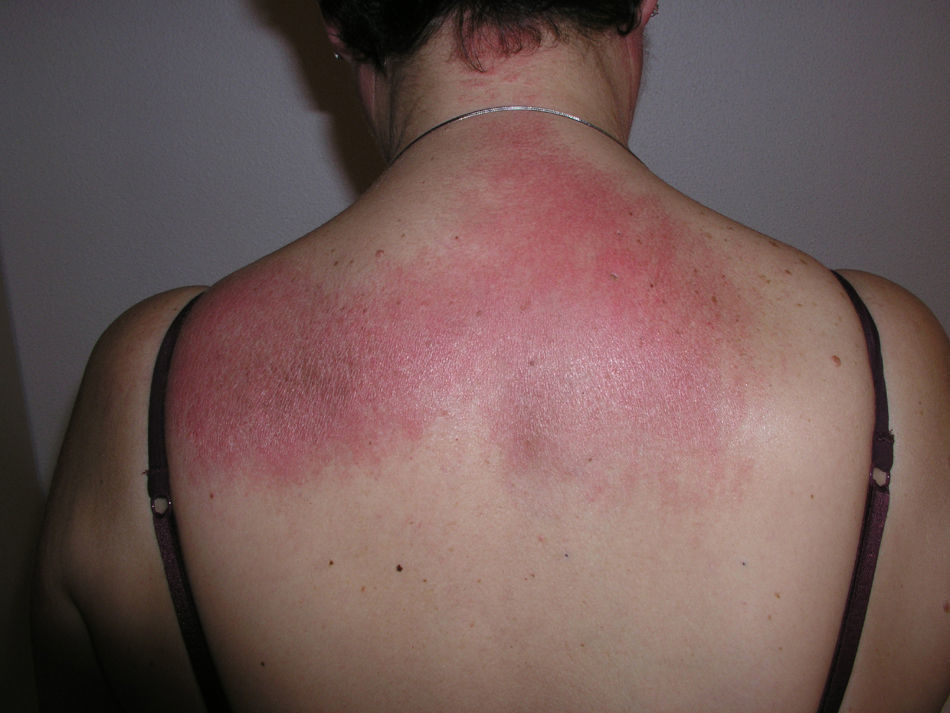
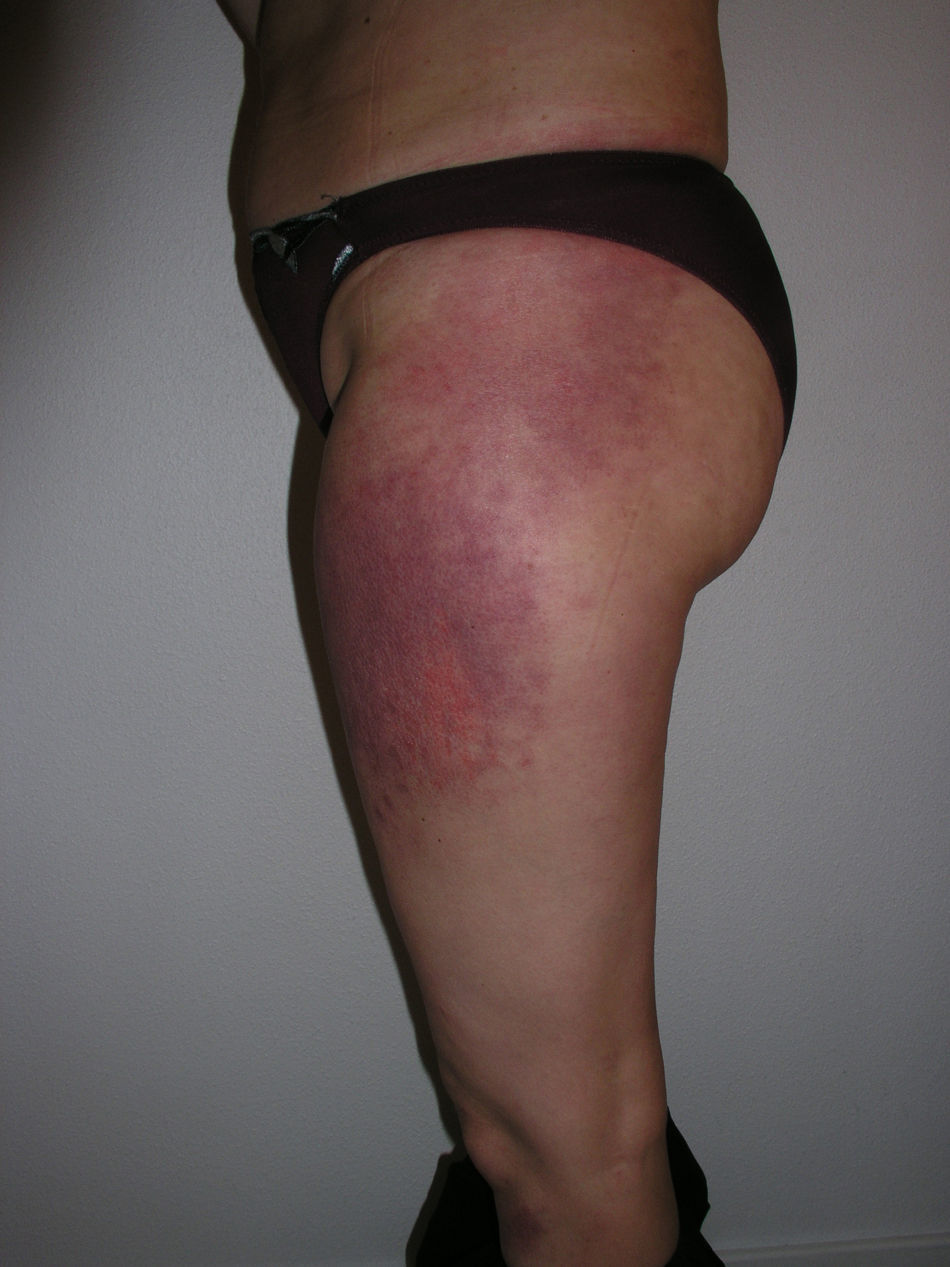
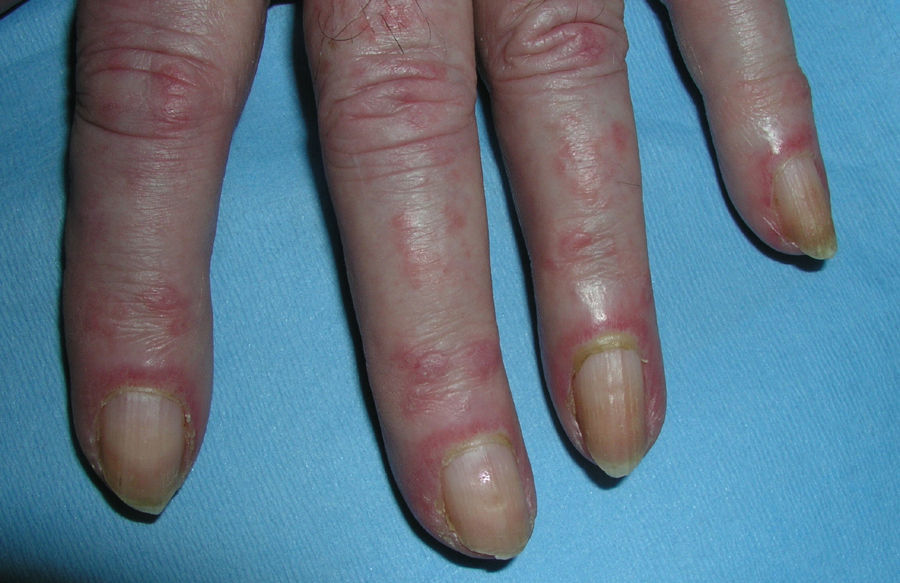
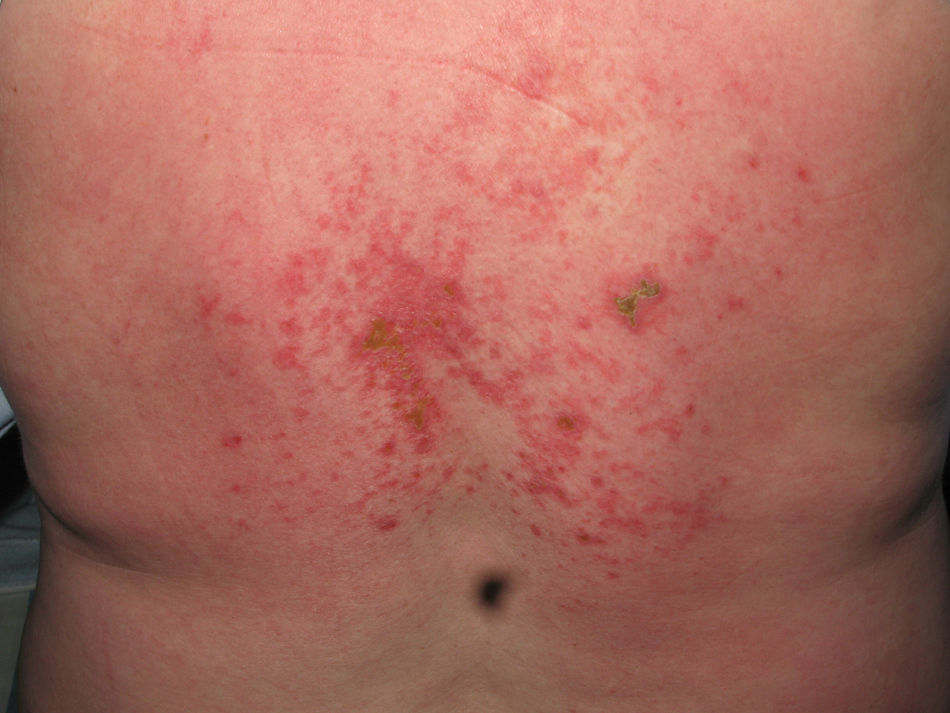
Skin Lesions in Patients With Dermatomyositis.
| Skin Lesions | Number of Patients | |
| Head and neck | ||
| Heliotrope erythema | 1,2,5,6,7,8,9,10,11,12,13,14,17,18 | |
| Violaceous erythema on the forehead and cheeks | 7,11,15,17,18 | |
| Psoriasiform dermatitis on the scalp | 5 | |
| Diffuse nonscarring alopecia | 11,16 | |
| Torso and limbs | ||
| V-sign on the chest | 1,2,5,6,9,10,11,12,17,18,20 | |
| Shawl sign | 5,6,9,10,20 | |
| Holster sign | 3,5,9,10,13 | |
| Violaceous erythema on the outer surface of the arms and on the elbows | 2,3,5,7,9,13,16,17,18,20 | |
| Necrotic lesions | 5,17,18 | |
| Hands | ||
| Gottron papules | 1,2,4,5,6,7,8,9,10,11,17,18 | |
| Periungual erythema | 1,4,6,7,9,11,12,13,14,15,17,18,19 | |
| Hypertrophic cuticles | 4,7,9,11,12,14,17,18,19 | |
| Calcinosis | 15,19 | |
| Necrosis of the fleshy parts of the fingers | 16 | |
| Others | ||
| Panniculitis | 3,6,7,16 | |
| Mechanic's hands | ||
| Poikiloderma |
Fifteen patients had generalized myalgia and proximal muscle weakness, 6 had arthralgia, 3 had dysphagia and dysphonia, and 2 developed diffuse interstitial lung disease. One of the patients with diffuse interstitial lung disease died after 4 months due to the lung disease.
Other ExaminationsHigh levels of enzymes associated with muscle damage (creatine kinase, aldolase, lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase) were detected in 15 patients. Antinuclear antibodies (ANA) were present in 6 patients. Analysis of myositis-specific antibodies, including anti-Jo1, anti-PL-12, anti-PL-7, anti-Mi-2, and anti-PRS was negative in all patients. Of the antibodies associated with myositis, anti-PM-Scl, anti-U1-RNP, and anti-SSA/RO were each present in 1 patient; negative results were obtained in tests for these antibodies in the other patients. Tumor-antigen markers tested for included CA19-9, CA125, PSA, CYFRA, CEA, and β-2 microglobulin. High levels of CA19-9 were detected in 2 patients (50 and 55.7 U/mL, respectively; normal values, <19 U/mL). β-2 microglobulin was found at a concentration of 4mg/L (normal values, <3.2mg/L) in 1 patient. A skin biopsy was performed on all patients and was compatible with a diagnosis of dermatomyositis in 18 patients. In 10 patients, biopsy of a Gottron papule showed variable hyperkeratosis with acanthosis and vacuolar degeneration of the basal layer. Vascular ectasia and a perivascular inflammatory infiltrate were visible in the dermis. Biopsy of the areas of violaceous erythema revealed interface dermatitis with vacuolar degeneration of the basal layer and epidermal atrophy in 8 patients. Histology revealed mucin deposits in the dermis of 11 patients. Electromyography showed a pattern of myopathic disease in 14 out of 18 patients. Muscle biopsy showed a pattern of myositis in 9 out of 12patients.
TreatmentInitial therapy was with systemic corticosteroids in 17 patients at a dosage of 0.5-1mg/kg/d for an average of 4 months. In 10 patients, therapy was continued with dosages of less than 0.5mg/kg/d for an average of 5 months. Patients undergoing treatment with systemic corticosteroids for more than 2 months developed a cushingoid habitus. It was necessary to add other drugs in 16 cases. Methotrexate was used in patients at a dosage of 15mg/wk, with a mean duration of 12 months. The drug was suspended in 1 patient due to gastric intolerance. Hydroxychloroquine was administered in 5 patients at a dosage of 400mg/d for an average of 14 months. Other treatments used are shown in Table 4.
Treatment in Patients With Dermatomyositis.
| Treatment | Dosage | Duration | Improvement | Adverse Effects | No. of Patients |
| Systemic corticosteroids | 0.5-1 mg/kg/d | 4 mo | Skin and muscle | Cushingoid habitus in 7 patients | 17 |
| < 0.5 mg/kg/d | 5 mo | 10 | |||
| Methotrexate | 15 mg/wk | 12 mo | Skin | Gastric intolerance in 1 patient | 14 |
| Azathioprine | 100 mg/d | 6 mo | Skin and muscle | - | 4 |
| Hydroxychloroquine | 400 mg/d | 14 mo | Skin | - | 5 |
| Chloroquine | 250 mg/d | 3 mo | Skin | Blurred vision in 1 patient | 1 |
| Thalidomide | 100 mg/d | 5 mo | Skin | - | 2 |
Four patients developed malignant tumors during the follow-up period. One patient had presented with endometrial adenocarcinoma 3 years before the diagnosis of dermatomyositis. The onset of clinical symptoms of dermatomyositis coincided with a relapse of the tumor. The patient died 3 months later due to disseminated metastases. Another patient was diagnosed with a small-cell lung carcinoma 2 months after onset of dermatomyositis. The patient responded well to cancer treatment and the skin lesions and muscle weakness improved at the same time. After 8 months of remission of the tumor, no evidence was found of skin or muscle disease. Another patient developed a parotid epidermoid carcinoma 18 months after the diagnosis of dermatomyositis was made. After 20 months of complete remission of the tumor the patient's skin lesions characteristic of dermatomyositis and the muscle weakness continued. A further patient was diagnosed with carcinoma in situ of the bladder. The skin lesions persisted after 24 months despite the fact that the patient was free from the tumor.
DiscussionIn our study, classic dermatomyositis was the most frequent presentation and was observed in 55% of patients. Amyopathic and cancer-associated dermatomyositis were observed in 15% and 10% of patients, respectively. These findings are similar to those reported in other studies.3,10,11 The disease was found to be predominant among women. A male-to-female ratio of 2:1 has been reported.1 This ratio was higher in our study, as only 1 male patient was included. However, the small number of patients in the study means that conclusions cannot be drawn regarding this point.
The most common skin lesions were heliotrope erythema and Gottron papules. More than half of the patients also had confluent macular violaceous erythema and periungual erythema. Violaceous erythema in areas exposed to sunlight and on extensor surfaces, together with lesions on the backs of the hands and around the fingernails should lead to suspicion of dermatomyositis. The most characteristic signs, which distinguish dermatomyositis from lupus erythematosus, are the violaceous appearance of the erythema and the tendency of the lesions to be distributed around the eyes and on extensor surfaces. Calcinosis cutis was observed on the fingers of 2 patients. One of these patients was diagnosed with juvenile dermatomyositis, consistent with the greater frequency of this finding in young patients.12 Necrotic lesions developed on the torso of 3 patients, 2 of whom had dermatomyositis associated with cancer. This type of lesion has been more commonly reported in patients with paraneoplastic dermatomyositis and should therefore increase the suspicion that the patient may have a malignant tumor.8,9
Arthralgia was considered to be mild in most cases. Two patients developed diffuse interstitial lung disease; in one of them the disease followed an acute course with severe dyspnea and hypoxemia, and the patient died 4 months later. In the other patient, the CT scan revealed ground glass opacity, but the patient remained asymptomatic throughout the follow-up period. Lung involvement has been reported in up to 40% of patients with dermatomyositis. These abnormalities are often only revealed in imaging studies.13–17 A further 3 patients presented dysphagia and dysphonia, which resolved rapidly during the first few months of treatment. Cardiac involvement has been reported in up to 50% of patients with dermatomyositis, but only a small proportion of these patients present symptoms.18 Gastrointestinal involvement is more common in the juvenile forms of the disease.19 No patients in our study had involvement of these organs.
Levels of enzymes that indicate muscle damage were elevated in most of the patients. These enzyme levels followed a random pattern and no trends were observed in any variable. Since some patients were already receiving treatment with corticosteroids when the first analyses were performed, some values may have been masked. All tests for myositis-specific antibodies were negative. These antibodies are considered highly specific to dermatomyositis, but their sensitivity is low and their absence cannot therefore rule out a diagnosis of this disease.5,20,21 Of the antibodies associated with myositis, anti-U1RNP and anti-SSA/RO were present in 2 patients with overlap syndrome. Anti-PM-Scl was present in 1 patient with amyopathic dermatomyositis. Three patients had slightly elevated levels of tumor markers. No tumor was detected in any of those patients after prolonged follow-up.
The prevalence of cancer in patients with dermatomyositis ranges from 9% to 42%.7–9,22 According to the Bohan and Peter criteria for dermatomyositis associated with cancer (tumor appears approximately 3 years after initial diagnosis of dermatomyositis and the dermatomyositis disappears when the tumor is eradicated), only 2 of the 4 patients with malignant tumors could be classified as patients with paraneoplastic dermatomyositis.6 Onset of dermatomyositis in the first patient coincided with a recurrence of the tumor and the patient died a few months later. In the second patient, the signs and symptoms of dermatomyositis practically disappeared when the cancer treatment finished. These 2 cases did follow a paraneoplastic course. In the other 2 patients, however, the characteristic dermatomyositis lesions persisted despite the long period of tumor remission. Early diagnosis of dermatomyositis and performing the extension study probably contributed to earlier diagnosis of the tumors. The identification of predictive factors for malignancy in adult dermatomyositis will make it possible to more accurately select those patients in whom an exhaustive search for a tumor is of the highest priority. Independent risk factors for an underlying tumor have been reported, including being over 52 years of age at the time of diagnosis, an abrupt and rapid onset of the skin lesions and muscle weakness, necrotic lesions and periungual erythema, and low levels of complement C4. Other factors present at higher frequencies in these patients are high levels of creatine kinase (>1000 U/L) and high titers of p155/140 antibodies. A low lymphocyte count (<1500/mm3) has been considered to be a protective factor.8
There is currently no consensus on the type of examinations that should be performed in patients with dermatomyositis in order to rule out an associated tumor. A reasonable option for men would include a complete clinical examination, a routine laboratory analysis, tumor antigen markers, and a CT scan of the chest and abdomen. A mammogram and pelvic ultrasound examination should be performed in women. An endoscopy study of the intestinal tract would be indicated by the clinical signs and symptoms and the age of the patient.5,23,24
Symptoms of other forms of collagenosis are observed in 20% of patients with dermatomyositis.2 Other indicators of collagenosis are observed alongside the skin and muscle lesions in dermatomyositis associated with other connective tissue diseases. The 2 patients who suffered from this form of dermatomyositis had more extensive systemic disease and multiple skin lesions.
Dermatomyositis-like symptoms can also be induced by drug treatments. This is most commonly observed in patients receiving hydroxyurea to treat chronic myeloproliferative diseases. These patients present skin lesions with no muscle or systemic disease. As in our study, the skin rash may appear after many years of treatment.25
Systemic corticosteroids continue to be the treatment of choice in patients with dermatomyositis.11 There is a clear discrepancy between the response to treatment of the muscle and skin disease. In our patients, systemic corticosteroids were withdrawn gradually as muscle and systemic symptoms improved. Muscle symptoms improved significantly after the first month of treatment. However, the skin lesions persisted for more than 6 months in more than half of the patients. Treatment of the skin manifestations continues to be unsatisfactory. In the first months of treatment, the tendency is to introduce drugs that reduce the need for corticosteroids.26,27 The most commonly used drug in our patients was methotrexate, followed by hydroxychloroquine. Skin lesions showed a good response to these treatments. A recent study showed that early use of methotrexate at a dosage of 15mg/wk made it possible to rapidly reduce the use of systemic corticosteroids and had fewer adverse effects.27 We used azathioprine in patients who were refractory to or had a poor tolerance of methotrexate, with partial results. Thalidomide was used in 2 patients who were refractory to the previous treatments, with good tolerance and a good response of the skin lesions. However, there is, as yet, no validated protocol for the treatment of dermatomyositis.
The data reported in our study are similar to those of previous studies, with the exception of the marked predominance in female patients. The characteristic skin lesions, the findings of the skin and muscle biopsies, and the electromyography results were conclusive for the diagnosis in most cases. Prognosis was favorable except in the cases associated with severe lung disease and those associated with a malignant tumor. The result of treatment was good in most cases, although long periods of treatment were required. Patients with paraneoplastic dermatomyositis were over 50 years of age, experienced a rapid onset of the clinical signs and symptoms of dermatomyositis, and had necrotic skin lesions. In these cases, a more exhaustive search for an associated tumor is recommended.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as Ciudad-Blanco C, et al. Dermatomiositis: estudio y seguimiento de 20 pacientes. Actas Dermosifiliogr.2011;102:448-455.


