




INTRODUCCIÓN
La criptococosis es una infección oportunista causada por Cryptococcus neoformans , un hongo encapsulado, que se encuentra en excrementos de aves y murciélagos, frutas, vegetales y en la tierra 1,2 . La principal puerta de entrada del agente infeccioso es la vía respiratoria. En los pacientes inmunocompetentes, habitualmente la infección se limita a esta localización, desarrollando, en algún caso, una sintomatología y una traducción radiológica escasa con resolución espontánea. En los pacientes con inmunosupresión celular (síndrome de inmunodeficiencia adquirida [SIDA], leucemia o linfoma, trasplantados y pacientes tratados durante un largo periodo de tiempo con esteroides y/o inmunosupresores) la infección puede diseminarse por vía hematógena y afectar a otros órganos, principalmente el sistema nervioso central (SNC) y la piel 3 . Las lesiones cutáneas secundarias aparecen entre el 10 y el 20 % de los casos, entidad conocida como criptococosis cutánea secundaria (CCS) 4,5 .
La criptococosis cutánea primaria (CCP) suele afectar a pacientes inmunodeprimidos, tras inoculación directa. Su diagnóstico se basa en la identificación de
C.neoformans en una biopsia o cultivo de la lesión cutánea, asociada a criterios clínicos (la existencia de un síndrome chancriforme) o histológicos (lesiones confinadas a la piel y el tejido celular subcutáneo), ambos con ausencia de diseminación, considerando que la linfadenopatía regional no se considera criterio de diseminación6 .
Se presenta un paciente con leucemia linfática crónica tipo B que desarrolló una CCP asociada a un fenómeno de oncotaxia.
DESCRIPCIÓN DEL CASO
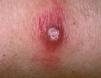
Un varón de 72 años presentaba, desde 1998, una leucemia linfática crónica (LLC) tipo B en estadio I que progresó a estadio III asociada a hipogammaglobulinemia. Desde su detección recibió tratamiento con 13 ciclos de clorambucilo y esteroides y, posteriormente, tres ciclos de ciclofosfamida, vincristina y prednisona hasta enero de 2002, asociados a gammaglobulina, con respuesta parcial. En marzo de 2002 el paciente presentó, en la cara anterior del muslo izquierdo, una lesión tuberosa de 2 meses de evolución, del color de la piel normal, asintomática, que había ido aumentando lentamente de tamaño, ulcerándose en su centro y adquiriendo una coloración vinosa en los bordes (fig. 1). No refería síntomas sistémicos acompañantes ni antecedentes de riesgo como traumatismo local o contacto con aves. Había realizado tratamiento antibiótico sistémico sin mejoría. En la exploración física no presentaba adenopatías regionales ni focalidad neurológica.

Fig. 1. Lesión tuberosa en el muslo.
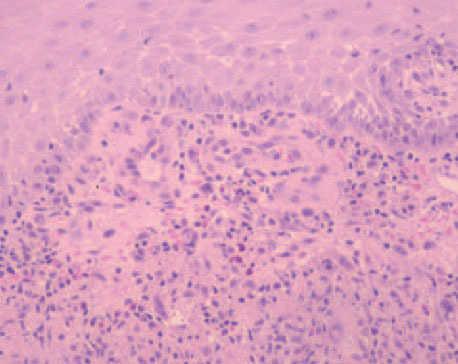
Fig. 2. Reacción inflamatoria granulomatosa. (Hematoxilina-eosi-na, ×100.)
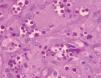
El estudio histopatológico de la lesión mostró una extensa ulceración epidérmica. En la dermis subyacente se observaba una extensa necrosis que se extendía al tejido celular subcutáneo, bordeada por una reacción inflamatoria granulomatosa con abundantes histiocitos, células gigantes multinucleadas, linfocitos, neutrófilos y eosinófilos (fig. 2). Tanto en el centro necrótico como en el borde granulomatoso se reconocían numerosos criptococos intracelulares y extracelulares (fig. 3). Así, se identificaban estructuras levaduriformes rodeadas por una gruesa cápsula ópticamente positiva con el azul alcián. En uno de los extremos podían distinguirse unos infiltrados de células linfoides de estirpe B, de aspecto maduro, monomorfas e hipercromáticas, CD20 y CD23 positivas, correspondientes a un infiltrado de células leucémicas (fig. 4). En el cultivo para hongos de la lesión se aisló C. neoformans .
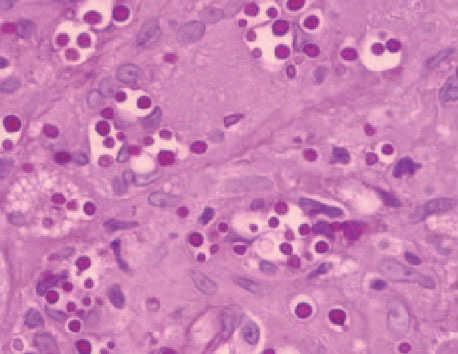
Fig. 3.Criptococos intracelulares y extracelulares. (PAS, ×400.)
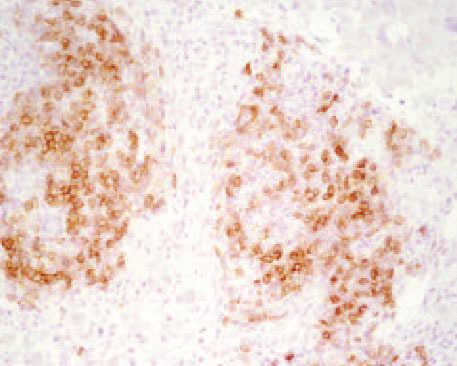
Fig. 4. Infiltrado nodular de linfocitos B (CD23+), compatible con oncotaxia. (Inmunoperoxidasa, ×200.)
En las pruebas complementarias destacaban leucocitos (10.390/l) con linfocitosis (55,80 %). El 49 % de las células presentaban marcadores de estirpe B (CD20, CD5, CD23) y el 36 % de estirpe T (CD4); plaquetas, 96.000/l; bilirrubina total, 1,40 mg/dl; transaminasa glutamicooxalacética (GOT), 44 U/l; lacticodeshidrogenasa (LDH), 626 U/l; e hipogammaglobulinemia. La serología para el virus de la inmunodeficiencia humana (VIH) era negativa. La tomografía computarizada (TC) toracoabdominal mostró numerosas adenopatías axilares, mediastínicas, mesentéricas, retroperitoneales e ilíacas y hepatoesplenomegalia. La aglutinación con látex en suero para la detección de antígenos de C. neoformans resultó negativa.
Con el diagnóstico de CCP, el paciente fue tratado con 400 mg/día de fluconazol durante 2 meses, con lo que la lesión involucionó y dejó una hiperpigmentación residual local. En diciembre de 2002 se observó en la TC abdominopélvica un aumento de tamaño de las adenopatías y de la esplenomegalia confirmándose un empeoramiento de la LLC sin observar recidiva de la CCP. El paciente falleció en enero de 2003 por insuficiencia cardiaca.
DISCUSIÓN
La existencia de la CCP como entidad ha sido muy debatida. En 1971 se consideró como «centinela de la infección diseminada»7 y en 1972 Noble y Fajardo8 , revisando 5 casos de esta patología, concluyeron que no podía existir sin asociarse a una previa, concomitante o posterior, diseminación sistémica.
Con posterioridad se ha documentado bien la criptococosis como una complicación de enfermedades que cursan con inmunodeficiencia celular, describiéndose casos de CCP en trasplantados renales6,9 y hepáticos10 , bajo terapia inmunosupresora, VIH posi-tivos/SIDA6,11-13 , linfopenia idiopática persistente CD4+5 , largos periodos de tratamiento esteroideo6,14 , linfoma no hodgkiniano y otras enfermedades malignas en tratamiento quimioterápico6,15 . Cabe destacar que la coexistencia de la CCP y enfermedades linfoproliferativas es inusual, ya que éstas cursan con inmunodeficiencia humoral. De hecho, en la revisión realizada de la literatura médica hemos encontrado, asociados a LLC, únicamente 2 casos de criptococosis diseminada16,17 y un caso de CCP asociado a traumatismo previo en la zona6 .
Las lesiones cutáneas pueden preceder en meses a las manifestaciones sistémicas, de forma que es importante establecer el diagnóstico diferencial entre CCP y CCS. Existen algunos criterios orientativos al respecto; en la CCP suele existir un antecedente traumático o un contacto previo con aves, la mayoría de las lesiones cutáneas son únicas, localizadas en zonas descubiertas y se presentan en forma de celulitis, úlceras, abscesos, lesiones tuberosas o eczematosas. En la CCS no existen los antecedentes descritos, las lesiones cutáneas son múltiples, de localización dispersa y muy polimorfas, destacando las lesiones acneiformes, placas infiltradas, nódulos, úlceras, celulitis, abscesos, lesiones tipo molusco contagioso, tipo pioderma gangrenoso o tipo sarcoma de Kaposi4,6 .
Histopatológicamente, se han descrito dos patrones de afectación cutánea dependientes del estado inmunitario del paciente y del hongo: la reacción gelatinosa y la granulomatosa. En la primera de ellas se observan masas de organismos rodeados por halos claros que representan la cápsula no teñida por la he-matoxilina-eosina y una escasa respuesta tisular. La segunda se caracteriza por presentar un menor número de microorganismos entremezclados con un infiltrado inflamatorio constituido por histiocitos, células gigantes, linfocitos y fibroblastos. La reacción tisular depende de la duración de la infección y no permite diferenciar la CCP de la CCS. La cápsula se tiñe de color púrpura con azul de metileno, de azul con azul alcián y de rojo con mucicarmina14,18 . El interés de nuestro caso radica en que, además de una reacción granulomatosa, se hallaron células linfoides de estirpe B, con marcadores CD20 y CD23 positivos, características de la LLC, secundario a un fenómeno de oncotaxia. Este fenómeno consiste en la presencia de células leucémicas en un foco inflamatorio. Su patogenia es desconocida, aunque podría obedecer a una respuesta inmunológica inespecífica a factores quimiotácticos liberados en este foco19 . Así, las células neoplásicas desaparecerían al resolverse el proceso inflamatorio con el tratamiento antifúngico sin precisar, por lo tanto, tratamiento quimioterápico específico, como de hecho ha sucedido con nuestro paciente.
En los pacientes con SIDA está bien establecido el tratamiento de la criptococosis sistémica. En la CCP no hay consenso, aunque los compuestos azólicos, como el fluconazol, pueden administrarse por vía oral y proporcionan una concentración tisular excelente y una actividad antifúngica similar a la anfoterizina B 20 , sin su toxicidad.
Correspondencia:
Marta Ballestero. Servicio de Dermatología. Hospital Universitario de La Princesa. Diego de León, 62. 28006 Madrid. España. agarcia@aedv.es
Recibido el 18 de mayo de 2004. Aceptado el 8 de julio de 2004.


