








El angiosarcoma primario cutáneo es uno de los tumores más agresivos y de peor pronóstico de la piel. Su clínica inicialmente indolente justifica frecuentes diagnósticos tardíos, lo que sumado a su carácter muchas veces multifocal y a su mala delimitación suele dificultar la cirugía. Debido a su baja frecuencia existen pocas series largas de casos tratados en un mismo centro. Revisamos las características clínico-patológicas de los angiosarcomas cutáneos tratados en nuestro centro en búsqueda de factores pronósticos, así como de posibles rasgos que faciliten un diagnóstico precoz.
Material y métodosSe realizó un estudio observacional retrospectivo de todos los pacientes diagnosticados de angiosarcoma cutáneo atendidos en nuestro hospital entre enero de 2000 y diciembre de 2015. Se recogieron 16 parámetros clínicos incluidos —entre otros— edad, sexo, tipo de angiosarcoma, localización, tamaño, tiempo de evolución y además 8 parámetros histopatológicos.
ResultadosSe recogieron 16 pacientes con angiosarcoma cutáneo —11 mujeres y 5 varones—, la media de edad fue de 67 años y la mediana de 71 años. La localización más frecuente fue el tronco con 10 casos, seguida de la cabeza y el cuello con 5 casos. La media del tamaño tumoral fue de 10cm y la mediana de 6,5cm. Se realizó escisión quirúrgica del tumor a 14 pacientes. Tras una media de seguimiento de 42,5 meses, 6 de los 16 pacientes seguían vivos al finalizar el estudio.
ConclusionesLa supervivencia de los pacientes con angiosarcoma cutáneo viene determinada principalmente por el tamaño tumoral y la edad. Otros rasgos asociados a peor pronóstico en nuestros pacientes fueron la infiltración a planos más profundos (músculo), un patrón histológico predominantemente sólido y un mayor número de mitosis.
Primary cutaneous angiosarcoma is one of the most aggressive skin tumors and carries a very poor prognosis. Its initially indolent clinical presentation explains the frequently late diagnosis that, together with its typically multifocal pattern and poor delimitation, often makes surgery difficult. The low incidence of primary cutaneous angiosarcoma means that few large single-center series have been published. We review the clinical and pathologic characteristics of cutaneous angiosarcomas treated in our hospital, looking for prognostic factors and for possible diagnostic traits that could facilitate early diagnosis.
Material and methodsThis was a retrospective observational study including all patients diagnosed with cutaneous angiosarcoma in Instituto Valenciano de Oncología in Valencia, Spain between January 2000 and December 2015. We recorded 16 clinical parameters, including age, sex, type of angiosarcoma, site, size, and time since diagnosis, and 8 histopathologic parameters.
ResultsWe identified 16 patients (11 women and 5 men) with cutaneous angiosarcoma. Their mean age was 67 years (median, 71 years). The most common site was the trunk (10 cases), followed by the head and neck (5 cases). The mean size of the tumor was 10cm (median, 6.5cm). Fourteen patients underwent surgical excision. Six of the 16 patients were alive at the end of the study, after a mean follow-up period of 42.5 months.
ConclusionsThe major determinants of survival among patients with cutaneous angiosarcoma are tumor size and patient age. Other characteristics associated with a poor prognosis were infiltration of deep planes (muscle), a predominantly solid histologic pattern, and a larger number of mitoses.
El angiosarcoma (AS) cutáneo es una de las neoplasias cutáneas de peor pronóstico. Su comportamiento es muy agresivo, con gran tendencia a la recidiva local y con una supervivencia a 5 años de entre el 12% y el 34% según la mayoría de los estudios1,2, aunque puede ser de hasta el 62%3. A diferencia de otros sarcomas, clásicamente el grado de diferenciación de los AS cutáneos no se ha relacionado con el pronóstico de estos tumores4.
La forma clásica de AS cutáneo es una lesión contusiforme, edematosa y mal definida, poco expresiva clínicamente en sus fases iniciales, que asienta en la cara o el cuero cabelludo de pacientes ancianos (AS de Wilson Jones) y que supone aproximadamente el 50% de los AS primarios cutáneos5–8. Además, existen otras 2 formas de presentación típica en la piel: el AS que asienta sobre áreas de linfedema crónico, especialmente en los brazos de mujeres sometidas a mastectomías radicales —conocidos como síndrome de Stewart-Treves9–11— y el AS posradioterapia que se desarrolla sobre áreas de piel irradiada, especialmente en la región pectoral de mujeres sometidas a radioterapia por un cáncer de mama12–15.
Su apariencia histopatológica varía desde formas relativamente diferenciadas, con luces vasculares reconocibles recubiertas por endotelios prominentes con algún grado de atipia y con un patrón infiltrativo, disecando los haces de colágeno, hasta formas muy indiferenciadas más sólidas compuestas por células fusiformes o epitelioides, mucho más atípicas, pleomórficas y con más mitosis, sin apenas luces vasculares y que ocasionalmente simulan carcinomas.
El tratamiento esencial del AS cutáneo y el único potencialmente curativo —si se consiguen márgenes libres— es la extirpación quirúrgica amplia seguida de radioterapia local, incluso algunos autores recomiendan irradiar los ganglios linfáticos regionales6. No obstante, en la mayoría de los casos conseguir márgenes libres no es fácil por la extensión del tumor, mucho más allá de lo que se aprecia clínicamente, y porque no es raro que se trate de tumores multicéntricos. La quimioterapia tiene solo un papel paliativo en el manejo de estos pacientes.
Aunque el AS cutáneo es un sarcoma muy poco frecuente —de hecho, los AS en su conjunto representan menos del 1% de todos los sarcomas— la mayoría de ellos se originan en la piel, pese a lo cual su escasa frecuencia justifica que muchos de los trabajos publicados de AS incluyan casos cutáneos junto con casos de origen visceral u óseo, que son aún de peor pronóstico que los cutáneos1. Esto motiva que escaseen los trabajos con un número considerable de casos en este campo, ya que es difícil conseguir series largas y uniformes de AS cutáneos2,4,5,7,16. Por otro lado, el manejo de estos pacientes es muchas veces desalentador, sobre todo en estadios avanzados con pronóstico infausto, pese a realizar tratamientos agresivos desde el inicio. Ambos hechos nos motivaron a recopilar los casos de AS cutáneos tratados en el Instituto Valenciano de Oncología en un intento de caracterizarlos clínica e histológicamente, así como de buscar, si los hubiera, factores clínicos, histológicos y terapéuticos que pudieran correlacionarse con el pronóstico. Así mismo, hicimos un esfuerzo especial en la revisión de las historias clínicas, así como de recuperación de los hallazgos clínicos, en búsqueda de datos exploratorios que pudieran servir de guía en el diagnóstico precoz de estos pacientes, ya que el diagnóstico en estadios tempranos y el pequeño tamaño mejoran sensiblemente las posibilidades de sobrevivir de estos pacientes.
Material y métodoSe realizó un estudio observacional retrospectivo de todos los pacientes diagnosticados de AS cutáneo atendidos en nuestro hospital entre enero de 2000 y diciembre de 2015. La fuente de recogida de todos los parámetros estudiados fueron las historias clínicas de los pacientes, el archivo de biopsias del servicio de anatomía patológica y el archivo fotográfico de nuestro servicio. En el estudio se incluyeron inicialmente 20 pacientes, de los cuales 4 tuvieron que ser excluidos. Los motivos de exclusión fueron 3: ausencia de seguimiento (en un caso), no disponer de material suficiente (en otro caso: no se pudo diferenciar entre hemangioendotelioma y AS), y no ser AS primarios cutáneos (en 2 casos). Estos 2 últimos casos habían sido etiquetados inicialmente como cutáneos porque en todos los cortes histológicos se apreciaba afectación cutánea de la mama por el AS. Posteriormente, al revisar los bloques, encontramos que la afectación cutánea era secundaria, de modo que ambos casos asentaban primariamente en el parénquima mamario desde el que se extendían secundariamente a la piel suprayacente.
En los criterios de inclusión se exigió una clínica sugestiva confirmada por una biopsia teñida con hematoxilina-eosina diagnóstica de AS, apoyada por estudios inmunohistoquímicos adicionales, que en la mayoría de los casos incluyó un CD31, CD 34, D2-40 y Ki-67.
Los parámetros estudiados en cada paciente fueron: edad, sexo, localización, tamaño, tipo de AS (primario, posradioterapia o poslinfedema), en los secundarios se recogió también el tipo de tumor anterior y los años transcurridos desde la radioterapia o el linfedema, el tipo de tratamiento del AS (cirugía, radioterapia, quimioterapia), recaída, metástasis, supervivencia y exitus. También se estudiaron datos histopatológicos que incluyeron: el estado de márgenes, el patrón histopatológico (vasoformativo, sólido o mixto), el tipo celular predominante (epitelioide, fusocelular), la necrosis (sí, no), el nivel de infiltración (epidermis, dermis, hipodermis, músculo, hueso), la reacción linfocitaria, la disposición del infiltrado y el número de mitosis en 10 campos.
ResultadosEn el estudio se recogieron un total de 16 AS cutáneos: 11 mujeres y 5 hombres, de edades comprendidas entre los 35 y los 83 años de edad, con una media de 67 años y una mediana de 71 años. La mayoría de nuestros casos correspondieron a AS posradioterapia (10 casos) seguidos del AS de Wilson Jones (5 casos) y solo un caso correspondió a un AS poslinfedema. La localización más frecuente fue el tronco (10 casos), seguida de la cabeza y el cuello (5 casos) y solo un caso se localizó en las extremidades superiores. El tamaño mínimo fue de 1cm y el máximo de 50cm, con una media de 10cm y una mediana de 6,5cm.
De los 11 casos que habían tenido un cáncer anterior, 10 tuvieron un cáncer de mama y uno un seminoma. De los cánceres de mama previos todos fueron carcinomas ductales infiltrantes salvo uno, que correspondió a un carcinoma lobulillar infiltrante.
En relación con los AS posradioterapia (n=10), la media de tiempo entre la aplicación de la radioterapia y la aparición del AS fue de 8,2 años. Casi todos los AS (9 casos) aparecieron tras al menos 5 años de la aplicación de la radioterapia y tan solo un caso apareció antes de los 5 años tras la misma.
En cuanto al tratamiento 14 casos fueron sometidos a cirugía, que se complementó con radioterapia en 4 casos. Ocho casos requirieron tratamiento quimioterápico, que solo en 2 casos fue el tratamiento inicial y único.
Con respecto al tipo de quimioterapia recibida, la doxorrubicina se empleó en 4 casos, el taxol también en 4 y la ifosfamida en 3, mientras que tanto el paclitaxel como la dacarbacina se utilizaron en un solo paciente cada uno. La respuesta a la quimioterapia fue pobre, de modo que aunque casi todos los casos tuvieron respuestas parciales, finalmente todos progresaron y fallecieron durante el seguimiento (8/8).
Se recogió la presencia de metástasis a distancia en 5 pacientes, de las cuales la mayoría fueron múltiples, y los lugares más frecuentes de asentamiento de estas metástasis fueron el pulmón y el hígado.
Diez de los 16 pacientes fallecieron por el AS durante el seguimiento. De los 6 casos restantes los 6 están actualmente libres de tumor. La media de seguimiento fue de 42,5, con una mediana de 26 meses y un rango de seguimiento de entre 7 y 188 meses.
En cuanto a los hallazgos histopatológicos el patrón fue sólido en 8 casos, en 4 fue vasoformativo y mixto en los otros 4. En relación con el tipo celular predominante 14 casos fueron predominantemente epitelioides y solo 2 predominantemente fusocelulares. Se observó necrosis en 6 AS y el nivel de infiltración fue subcutáneo en la mayor parte de casos (10 pacientes), 4 casos se confinaron a la dermis y solo 2 alcanzaron el plano muscular. Con respecto a los márgenes quirúrgicos hubo 3 casos no valorables, 8 casos en los que se obtuvieron márgenes negativos y 5 casos con márgenes afectos. La reacción linfocitaria del tumor fue escasa o moderada en 10 casos, intensa en otros 2 y nula en los 4 restantes. De los 14 casos en los que hubo alguna reacción linfocitaria dicho infiltrado se dispuso a nivel peritumoral en 2 casos, intratumoral en 8 y hubo 2 casos que tuvieron ambos. En cuanto a las mitosis encontramos una media de 15 mitosis por 10 campos con un rango de entre 0 y 37 mitosis.
Los resultados más relevantes clínicos y patológicos se resumen en la tabla 1. Los resultados del análisis comparativo entre los supervivientes y los fallecidos se resumen en la tabla 2.
Selección de resultados clínico-patológicos de los 16 angiosarcomas cutáneos
| Paciente | Edad | Sexo | Tipo | Localización | Tamaño (cm) | Tiempo Rx | Dosis Gy | Tumor previo | AP ca. de mama |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | M | Rt | Mama izq | 18 | 60 | 46 | Mama | Ca. ductal infiltrante |
| 2 | 71 | M | ST | Brazo izdo | 12 | – | - | Mama | CDI |
| 3 | 51 | M | Rt | Submama dcha | 1 | 57 | 50 | Mama | CDI |
| 4 | 76 | M | WJ | CC | 3 | – | – | No | . |
| 5 | 77 | M | Rt | Mama dcha | 1 | 94 | 46 | Mama | CDI |
| 6 | 71 | M | Rt | Mama izda | 50 | 171 | 48 | Mama | CDI |
| 7 | 48 | V | Rt | Pared abd | 2 | 96 | 26 | Seminoma | – |
| 8 | 55 | M | Rt | Mama izda | 10 | 88 | 46 | Mama | C. lobulillar infiltrante |
| 9 | 69 | M | Rt | Mama izda | 8 | 143 | 46 | Mama | CDI |
| 10 | 76 | V | WJ | Mejilla dcha | 6 | – | – | No | - |
| 11 | 35 | M | Rt | Mama dcha | 12 | 66 | 50 | Mama | CDI |
| 12 | 57 | M | Rt | Mama dcha | 8 | 108 | 50 | Mama | CDI |
| 13 | 68 | V | WJ | CC | 2 | – | – | No | - |
| 14 | 80 | V | WJ | CC | 15 | – | - | No | - |
| 15 | 79 | V | WJ | CC | 2 | - | - | No | - |
| 16 | 83 | M | Rt | Mama izda | 3 | 110 | 50 | Mama | CDI |
| X=67,1 | 11M 5V | 10 Rt, 5 WJ 1 ST | 9mama, 5 cc, abdomen, 1 EESS | X=10 | X=100,3 | X=45,8 | 10 mama Un seminoma | 10 ca. de mama: 9 CDI 1 CLI |
| Paciente | Cirugía | Margen (cm) | Tto | Exitus | Patrón AP | Tipo celular | Necrosis | Nivel infiltración | Mit. mm2 | Supervivencia (meses) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | S | 0,2 | 13 | Sí | 1 | E | N | 3 | 7 | 24 |
| 2 | N | nc | 3 | Sí | 2 | E | N | 3 | 37 | 8 |
| 3 | S | 3,5 | 1 | No | 1 | E | N | 3 | 2 | 29 |
| 4 | S | 2 | 123 | Sí | 2 | E | N | 3 | 28 | 26 |
| 5 | S | nc | 1 | Sí | 2 | E | N | 2 | 14 | 8 |
| 6 | N | nc | 3 | Sí | 1 | E | N | 2 | 0 | 19 |
| 7 | S | nc | 1 | No | 3 | E | N | 3 | 6 | 187 |
| 8 | S | nc | 13 | Sí | 2 | E | S | 4 | 6 | 28 |
| 9 | S | nc | 13 | Sí | 3 | E | S | 3 | 22 | 24 |
| 10 | S | nc | 12 | Sí | 2 | E | S | 3 | 16 | 7 |
| 11 | S | 0,5 | 1 | No | 2 | F | N | 3 | 18 | 76 |
| 12 | S | 0 | 13 | Sí | 3 | E | S | 3 | 36 | 26 |
| 13 | S | 2 | 12 | No | 3 | E | N | 3 | 5 | 95 |
| 14 | S | 2 | 123 | Sí | 2 | E | S | 4 | 17 | 24 |
| 15 | S | 2 | 1 | No | 2 | E | S | 2 | 6 | 53 |
| 16 | S | 3 | 1 | No | 1 | F | N | 2 | 23 | 51 |
| 14 sí, 2 no | 1,68 | 14:Qx 4: Rt 8: Qt | 10 sí 6 no | 4 vasof 8 sólid 4 mixto | 14 E, 2 F | 6 Sí 10 No | 4: dermis 10: hipod 2: músc | X=15 | X=42,8 |
En negrita los pacientes fallecidos.
AP: anatomía patológica (1-vasoformativo [vasof], 2-sólido [sólid], 3-mixto); CDI: carcinoma ductal infiltrante; D: derecho; E: epitelioide; EESS: extremidades superiores; F: fusocelular; I: izquierdo; N: no; nivel de infiltración (2 dermis, 3 hipodermis, 4 músculo); Rt: angiosarcoma posradioterapia; S: sí; ST: angiosarcoma de Stewart Treves; tiempo Rx: latencia desde la radioterapia (en meses); Tto: tratamiento (1 cirugía, 2 radioterapia, 3 quimioterapia); WJ: angiosarcoma de Wilson Jones; X= media.
Análisis comparativo de resultados entre el grupo de pacientes supervivientes y los fallecidos por angiosarcoma cutáneo
| Parámetros | Vivos (6 casos) | Fallecidos (10 casos) |
|---|---|---|
| Edad | 61 años | 71 años |
| Mujeres | 3 | 8 |
| Varones | 3 | 2 |
| Posradioterapia | 4 | 6 |
| Idiopático | 2 | 3 |
| Poslinfedema | 0 | 1 |
| Tronco | 4 | 6 |
| Cabeza y cuello | 2 | 3 |
| EESS | 0 | 1 |
| Tamaño (cm) | 3,6 | 13,1 |
| Latencia tras RT (meses) | 82,25 | 110,6 |
| Dosis RT (Gy) | 44 | 47 |
| Mama | 3 | 7 |
| Seminoma | 1 | |
| Cirugía | 6 | 8 |
| Radioterapia | 1 | 3 |
| Quimioterapia | 0 | 8 |
| Vasoformativo | 2 | 2 |
| Sólido | 2 | 6 |
| Mixto | 2 | 2 |
| Necrosis | 1 | 5 |
| Dermis | 2 | 2 |
| Subcutáneo | 4 | 6 |
| Músculo | 0 | 2 |
| Mitosis | 10 | 18,3 |
| Supervivencia (meses) | 81,8 | 19,4 |
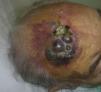
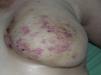
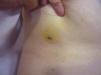
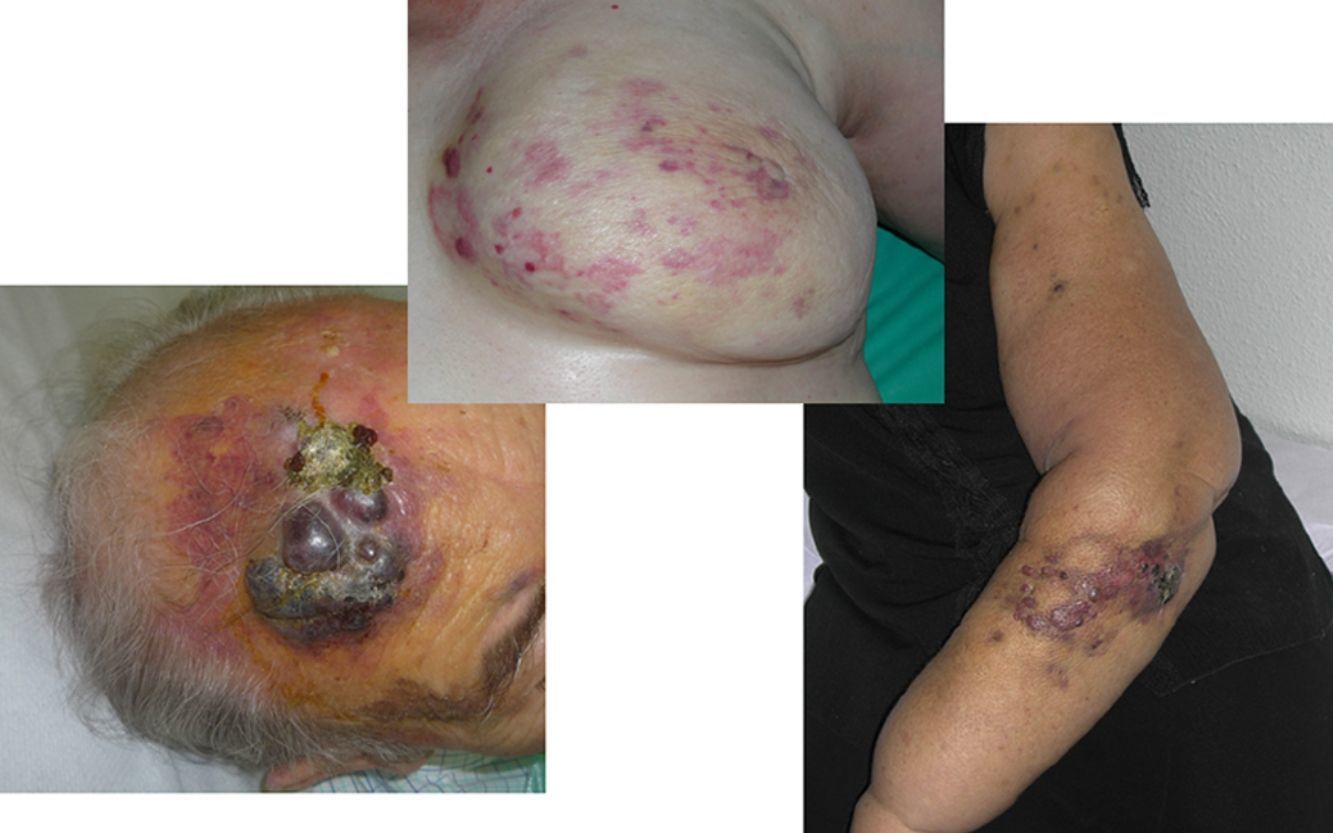
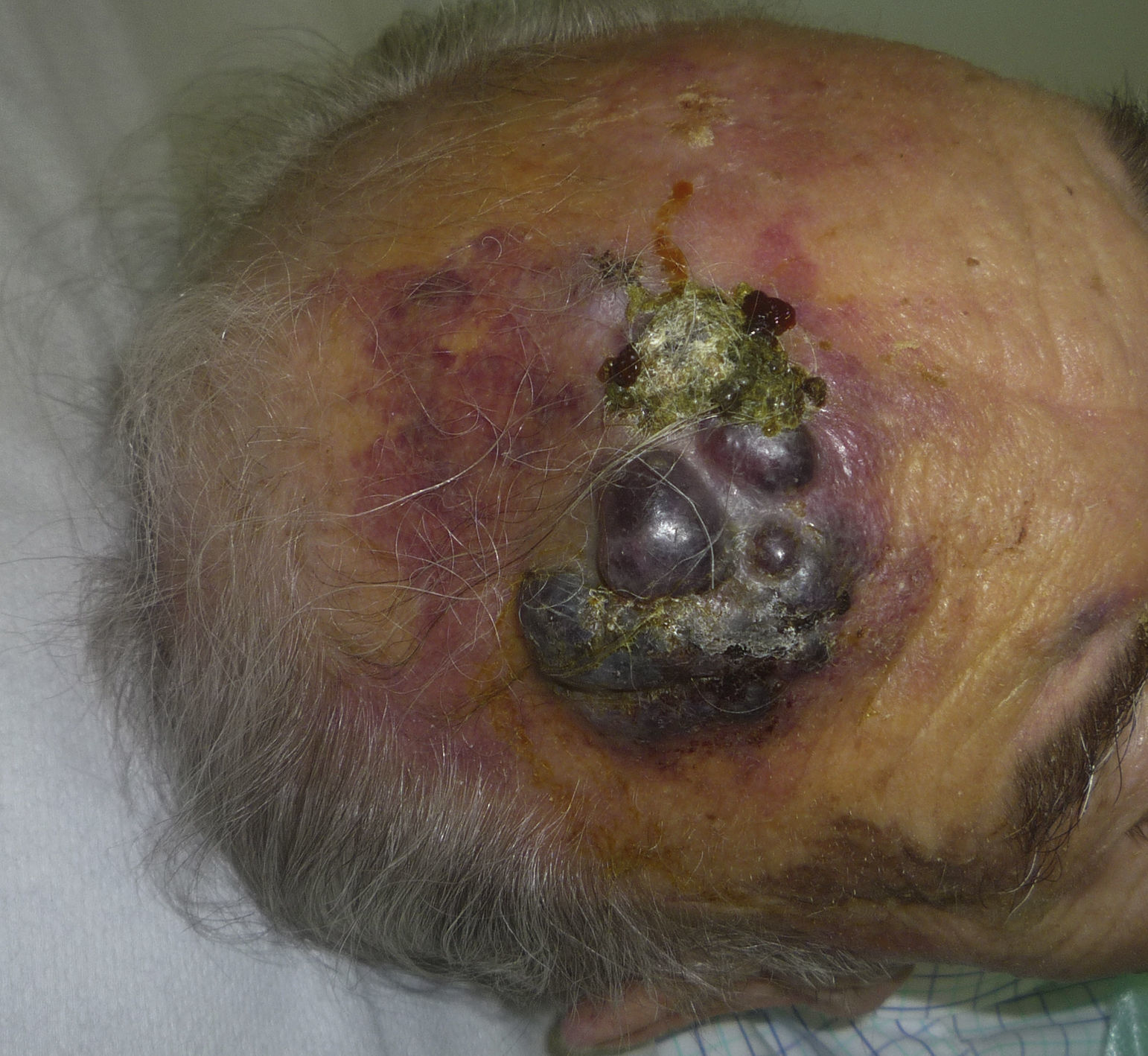
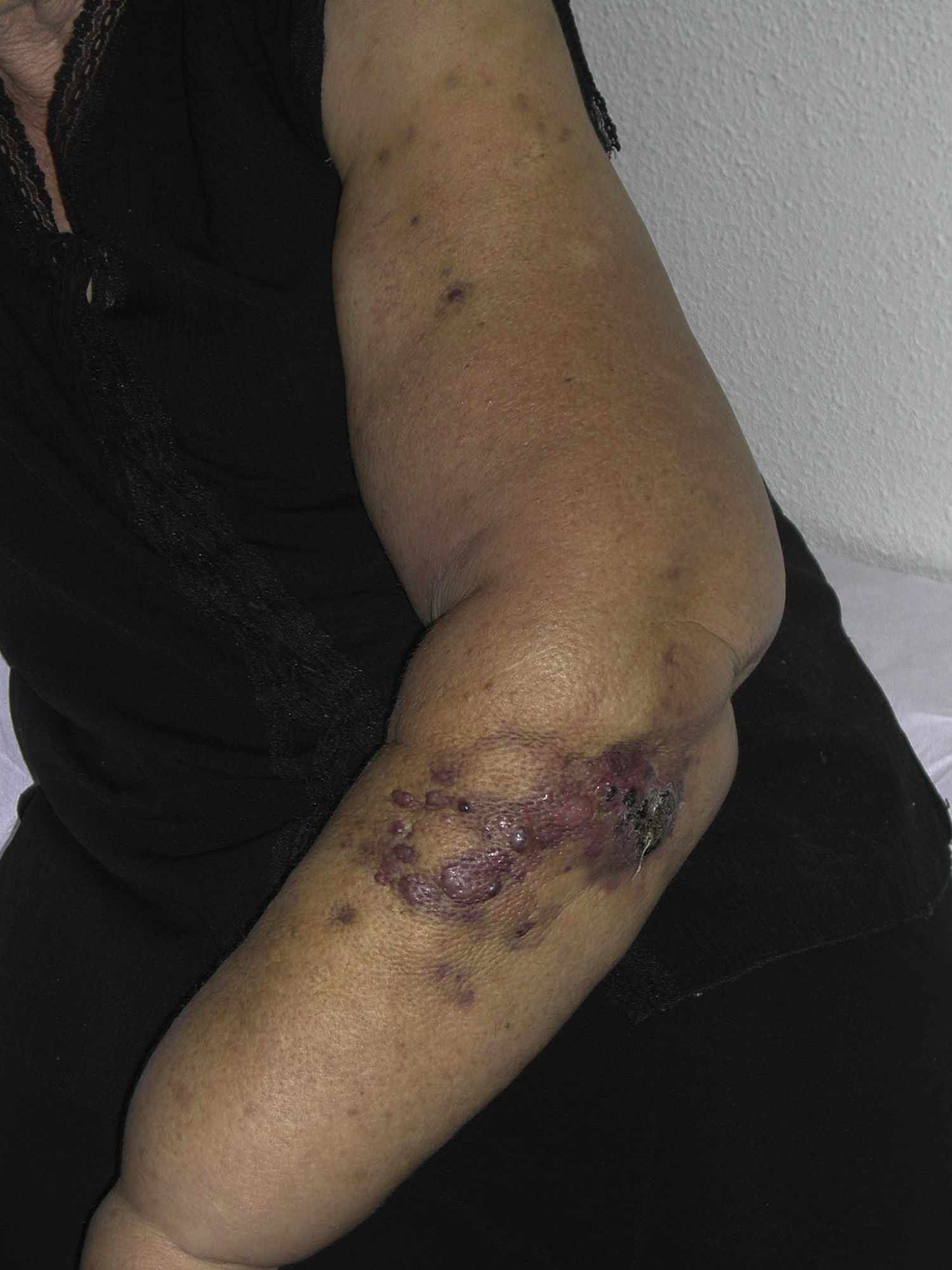
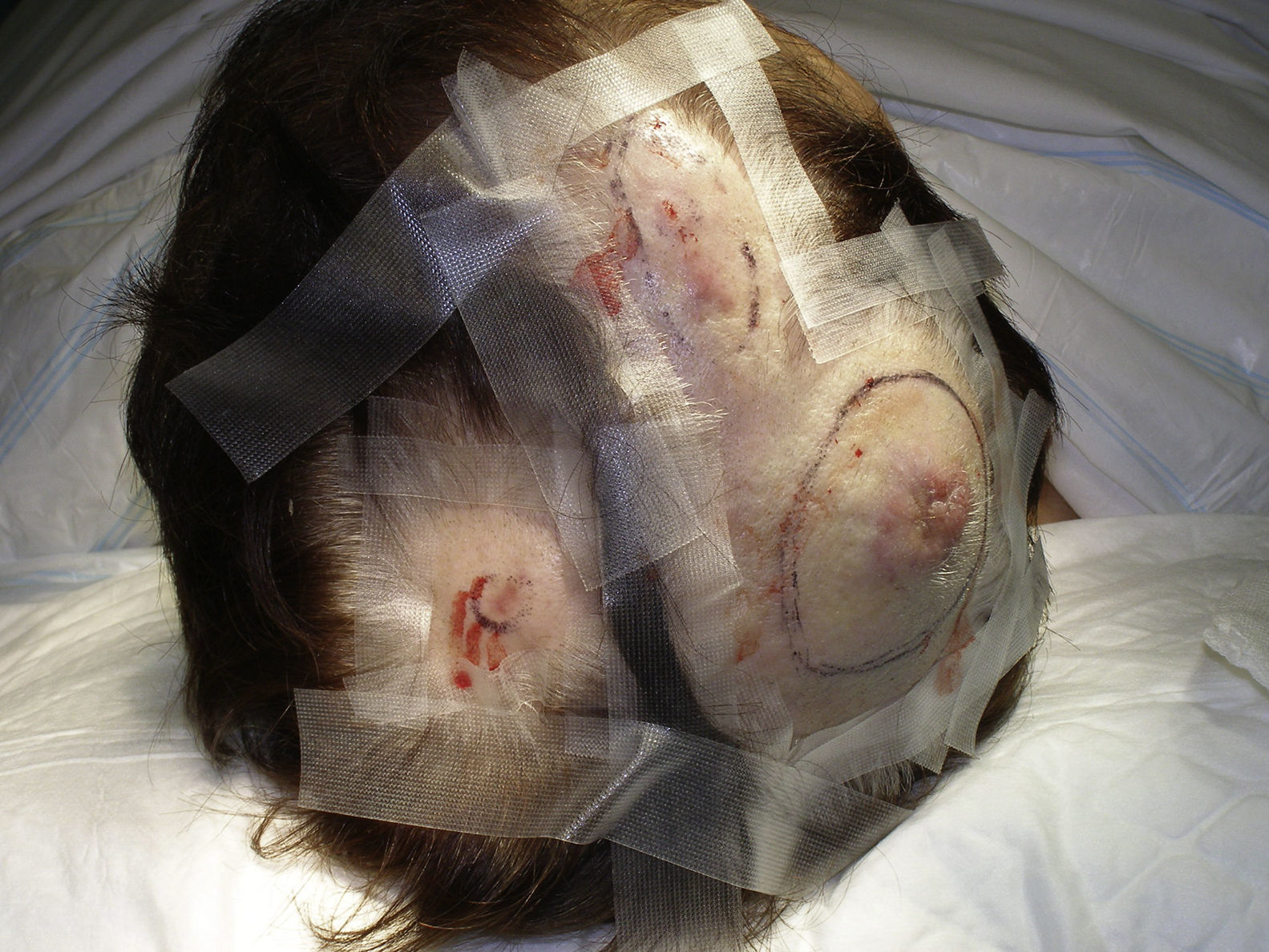
El AS cutáneo es un tumor muy poco frecuente, y el hecho de que en un centro exclusivamente oncológico solo hayamos sido capaces de recopilar 16 casos durante un periodo de 14 años lo corrobora. En su conjunto, el AS cutáneo es algo más frecuente en hombres de edad avanzada. Esto se debe a que la forma más frecuente de AS cutáneo en la población general es la del AS primario de cabeza y cuello o AS idiopático o de Wilson-Jones (fig. 1), que efectivamente suele afectar a varones ancianos5,7,17. Por su parte, el AS posradioterapia (fig. 2) es actualmente el segundo más frecuente debido al incremento del uso de la radioterapia en el cáncer de mama en decremento de las mastectomías radicales12,13,18, lo que ha condicionado a su vez una disminución en la frecuencia de los AS poslinfedema, que son los AS menos frecuentes en la actualidad. En nuestra serie, debido a la gran cantidad de mujeres con cáncer de mama que atiende el IVO, los resultados no coinciden con lo descrito en la literatura, y el tipo de AS cutáneo predominante fue el AS posradioterapia. Esto motiva también el predominio de mujeres en nuestra serie, en la que 11 de los casos fueron mujeres y solo 5 fueron hombres. Solo tuvimos un caso de AS poslinfedema (fig. 3), de modo que fue la variedad menos común, esta vez sí en consonancia con lo descrito. Se trataba de una mujer con un linfedema crónico del brazo izquierdo secundario a una linfadenectomía axilar por un cáncer de mama 22 años antes.
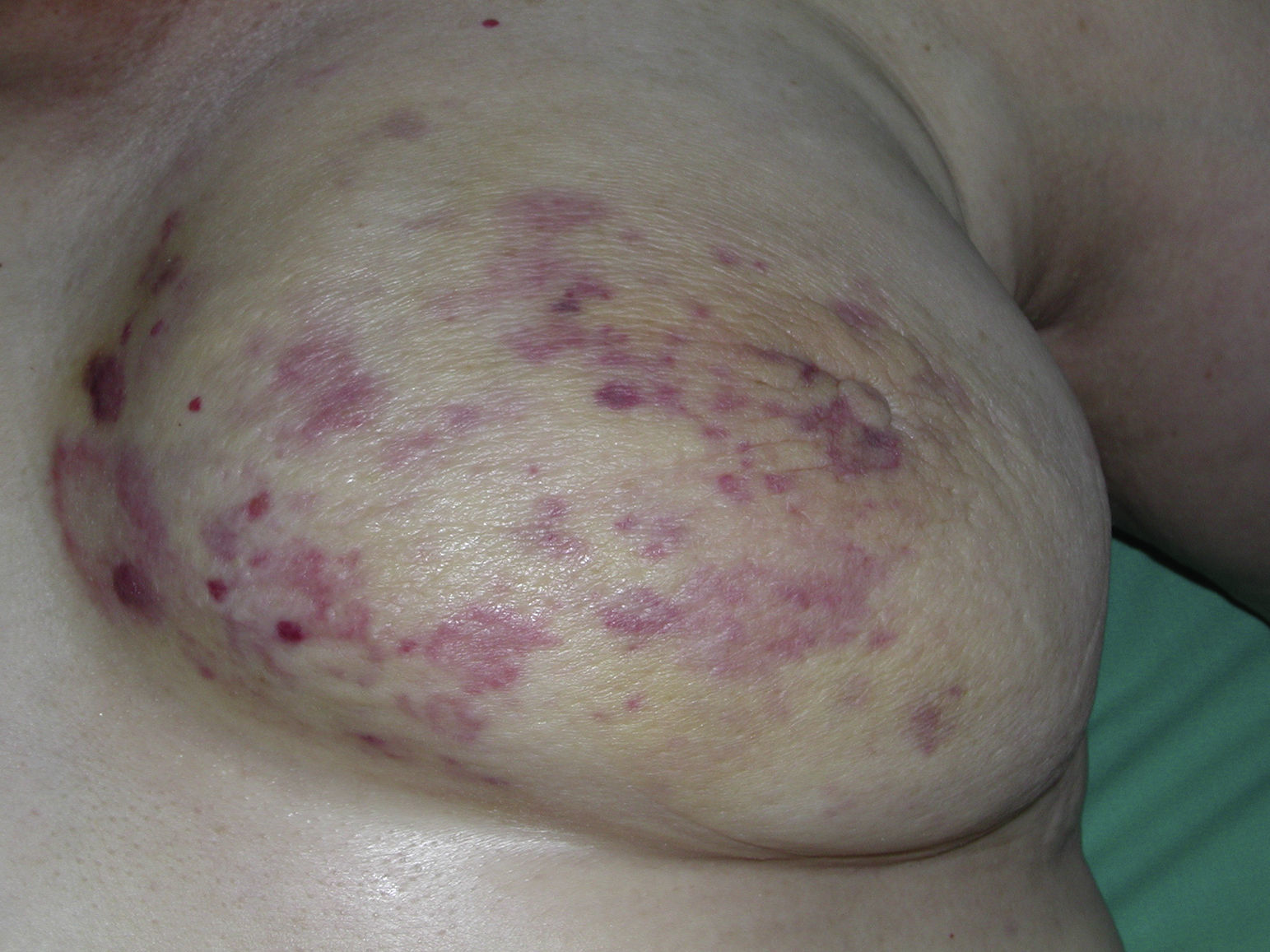
De esas 11 mujeres todas, salvo una que correspondió a un AS idiopático o de Wilson Jones, tuvieron un cáncer de mama previo. El 90% de dichos cánceres correspondieron a carcinomas ductales infiltrantes. Dado que en la población general el 80% de los cánceres de mama corresponden a carcinomas ductales, y solo un 10% a carcinomas lobulillares, probablemente no exista una especial asociación entre el AS cutáneo posradioterapia y un tipo de cáncer de mama específico, como podría desprenderse de nuestros resultados. Por el contrario, esta proporción refleja simplemente la ratio normal de los distintos cánceres de mama en la población. Dentro de este epígrafe del AS de localización mamaria cabría comentar una curiosidad de nuestro estudio. Está descrito que el AS de mama suele ser cutáneo si es radioinducido, y en cambio cuando no es radioinducido suele ser parenquimatoso19. Pues bien, de los casos recogidos inicialmente para este trabajo 2 correspondían a AS mamarios no posradioterapia. Tras revisar estos 2 casos constatamos que en ambos la localización primaria del tumor era el parénquima mamario, no la piel, de modo que la afectación cutánea era secundaria por infiltración posterior, por lo que fueron rechazados para este trabajo. El resto de los mamarios fueron todos primariamente cutáneos y radioinducidos, en consonancia pues con lo descrito.
La localización más frecuente del AS en el conjunto de nuestros casos fue la mama, con 10 casos en total, y no la cara y el cuero cabelludo, como sería esperable. El único caso localizado en una extremidad fue el caso poslinfedema. Nuevamente, la localización predominante se viene a explicar por la preponderancia de casos en nuestra serie de los AS posradioterapia de mama.
Aunque el tiempo de latencia entre la aplicación de la radioterapia y la aparición del AS es muy variable en la literatura (3-50 años), este periodo es más largo cuando la radioterapia es indicada por enfermedades benignas, con un tiempo de latencia de 25 años de media. En cambio, en los casos en los que la radioterapia se emplea sobre procesos patológicos malignos parece que el tiempo de latencia está en torno a los 10-15 años, salvo en el caso de la mama, en el que la media está en torno a los 5 años12. No está claro el motivo de esta latencia más corta en el caso de la mama, aunque las teorías propuestas incluyen el gran volumen de piel irradiada, un componente de linfedema asociado, posibles características intrínsecas de la mama y la posibilidad de una sinergia con la quimioterapia13,14. En nuestros 10 casos posradioterapia la media del tiempo de latencia entre la radioterapia y el AS fue de 8,2 años, con al menos 5 años de latencia en 9 casos. No está descrito en la literatura si el tiempo de latencia entre la aplicación de la radioterapia y el desarrollo del AS tiene alguna influencia en el pronóstico del AS. En nuestra serie este tiempo de latencia fue algo superior en el grupo de los fallecidos (110,6 meses) que en el grupo de los vivos (82,25 meses). En el único caso de síndrome de Stewart-Treves de nuestra serie el tiempo de latencia entre la linfadenectomía y el AS fue de 22 años, mientras que en la literatura el tiempo de latencia entre el desarrollo del linfedema y el AS es muy variable, de uno a 30 años, con una media de 10 años. El síndrome de Stewart Treves es la variedad más común de AS poslinfedema9,11, de modo que supone el 90% de los casos, pero existen también AS sobre linfedemas de otras etiologías, como linfedema congénito, linfedema por filariasis, linfedemas por adenoidectomías de otro origen, etc.10.
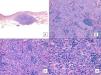
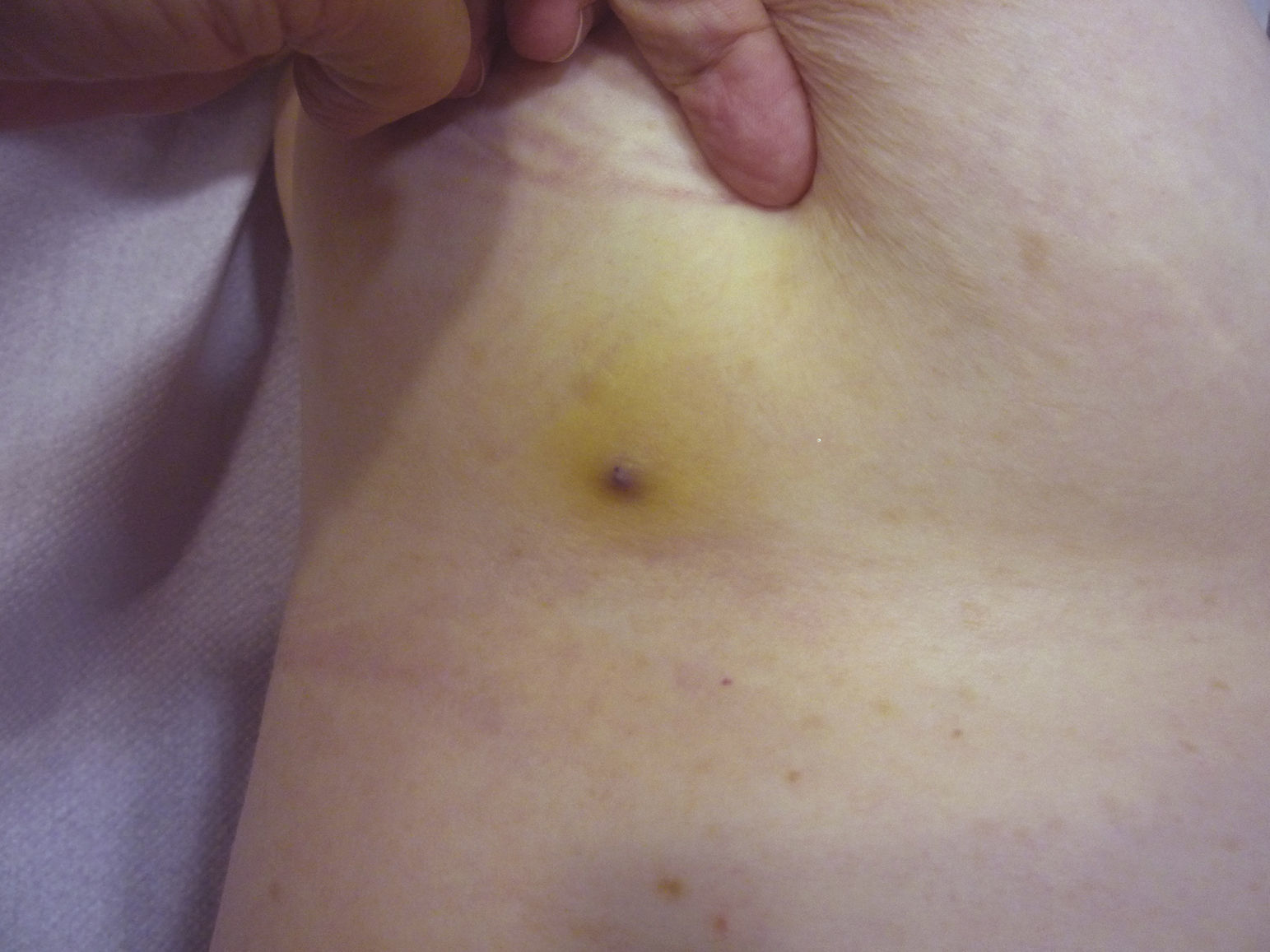
De los marcadores pronósticos el tamaño tumoral es actualmente el más aceptado, de modo que está bien documentado que tienen peor pronóstico los AS con un tamaño igual o mayor a 5cm que los menores de 5cm5,17. En nuestros casos encontramos diferencias en el tamaño medio entre el grupo de los supervivientes y el de los fallecidos. Así, en el grupo de pacientes supervivientes la media del tamaño fue de 3,6cm, en comparación con los exitus, en los que la media fue de más del triple, con 13,1cm. Por lo tanto, nuestros casos apoyarían que un mayor tamaño se asocia a un peor pronóstico del AS, como está descrito. Sería esencial conseguir diagnosticar los AS en sus fases iniciales, contusiformes, lo cual es especialmente difícil en los ASs de cuero cabelludo, más aún cuando este conserva el pelo (fig. 4). No es tan difícil en el caso del AS posradioterapia de la mama, donde cualquier lesión de apariencia vascular o contusiforme persistente debería ser biopsiada. Como clave diagnóstica puede ser de utilidad encontrar un halo amarillento en torno a la lesión sospechosa (fig. 5), que corresponde a hemosiderina, y que nosotros no hemos encontrado nunca en proliferaciones vasculares benignas sobre la piel irradiada. El diagnóstico diferencial entre el AS posradioterapia y las proliferaciones vasculares atípicas desarrolladas sobre piel irradiada se basa en rasgos histopatológicos, pero también son de ayuda algunos rasgos clínicos. Las proliferaciones vasculares atípicas sobre piel irradiada suelen ser de mucho menor tamaño que los AS, y el tiempo de latencia desde la radioterapia suele ser también menor20. A diferencia de los AS, las proliferaciones vasculares atípicas sobre piel irradiada se confinan a la dermis superficial y media y no alcanzan el tejido celular subcutáneo. Además, no se observa la atipia citológica nuclear de los endotelios, ni la presencia de múltiples capas de células endoteliales, ni las mitosis que se observan en los AS. Pese a todo, el diagnóstico diferencial es en ocasiones difícil y existen casos con coexistencia de ambos tipos de lesiones en una misma mama irradiada. Incluso existen casos documentados de proliferación vascular atípica posradioterapia evolucionados a AS21. En casos muy difíciles puede ser de utilidad realizar estudios en búsqueda de una posible sobreexpresión —si se emplea inmunohistoquímica— o amplificación —si se usa el FISH— del gen MYC. En efecto, parece que las proliferaciones vasculares atípicas sobre piel irradiada nunca muestran amplificación del MYC, que por el contrario, es bastante común en los AS22.
El otro marcador pronóstico más aceptado en la literatura es la edad, y en nuestro grupo fue menor en el grupo de los vivos (62 años) que en el de los fallecidos (71 años). Por otro lado, del conjunto de los 16 casos 10 habían fallecido al final del estudio, lo cual corrobora el mal pronóstico clásico que se le achaca al AS.
Con respecto al aspecto histológico, a diferencia de otros sarcomas la AJCC no considera el grado histológico relevante en el pronóstico de los AS. Pese a ello, algunos autores proponen posibles influencias pronósticas de algunos rasgos histopatológicos. Por ejemplo, según trabajos recientes, el predominio del patrón sólido sería un factor de relativo buen pronóstico en AS de cabeza y cuello3,16. Sin embargo, en nuestra serie esto no se cumple, ya que el patrón histológico sólido predominante se observó en 6 pacientes del grupo de fallecidos (6/10) y solo en 2 pacientes de los que permanecían vivos al final del estudio (2/6) (figuras 6 y 7).
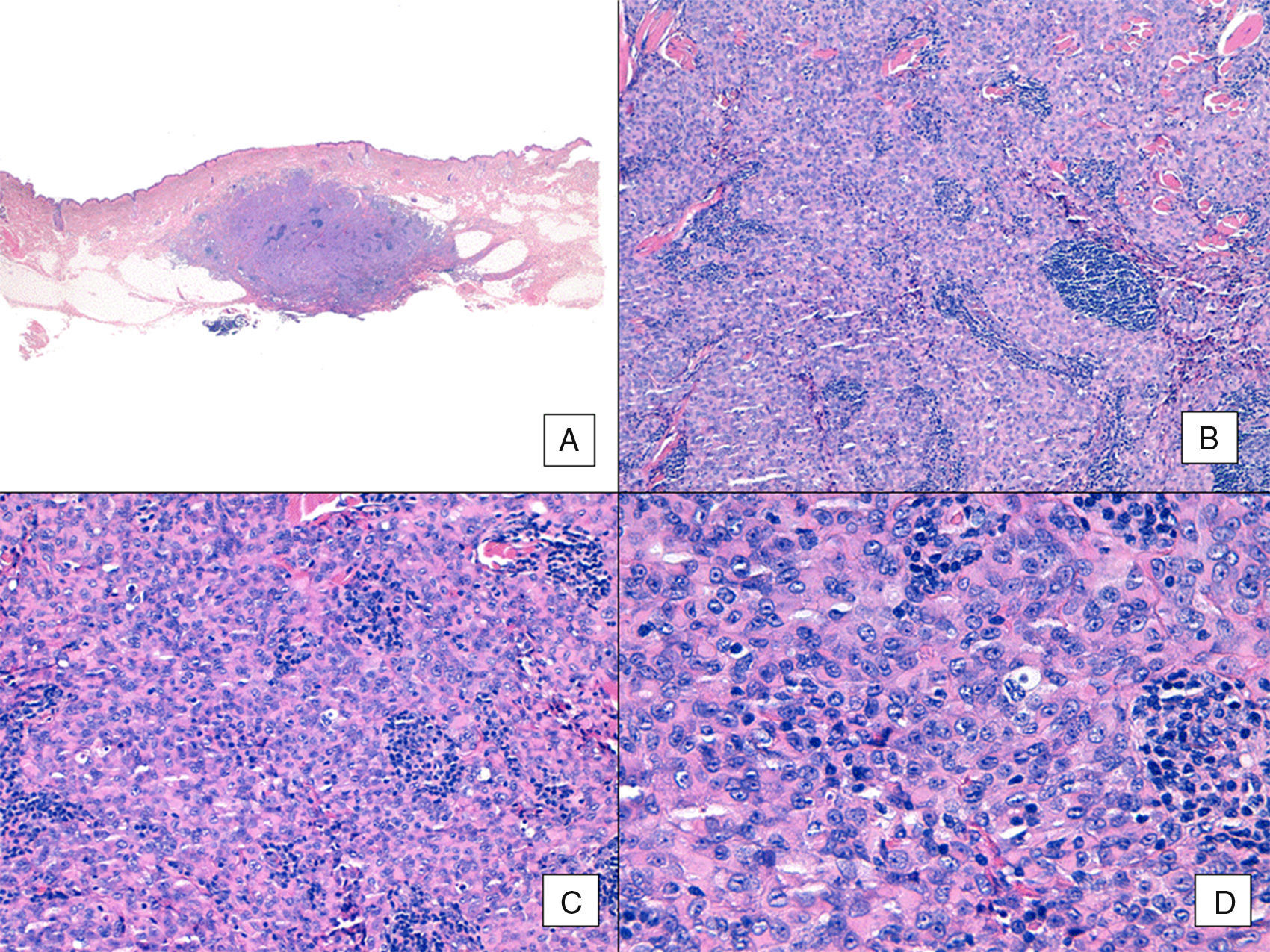
Angiosarcoma con predominio de patrón sólido. A. En la imagen panorámica el tumor infiltra la dermis reticular media y profunda y la hipodermis (hematoxilina-eosina ×10). B. El tumor es densamente celular, destruye las estructuras preexistentes y se acompaña de nódulos de infiltrado linfocitario (hematoxilina-eosina ×100). C y D. En el detalle se aprecia un predominio de células epitelioides acompañadas de infiltrado linfocitario (hematoxilina-eosina ×200 y ×400, respectivamente).
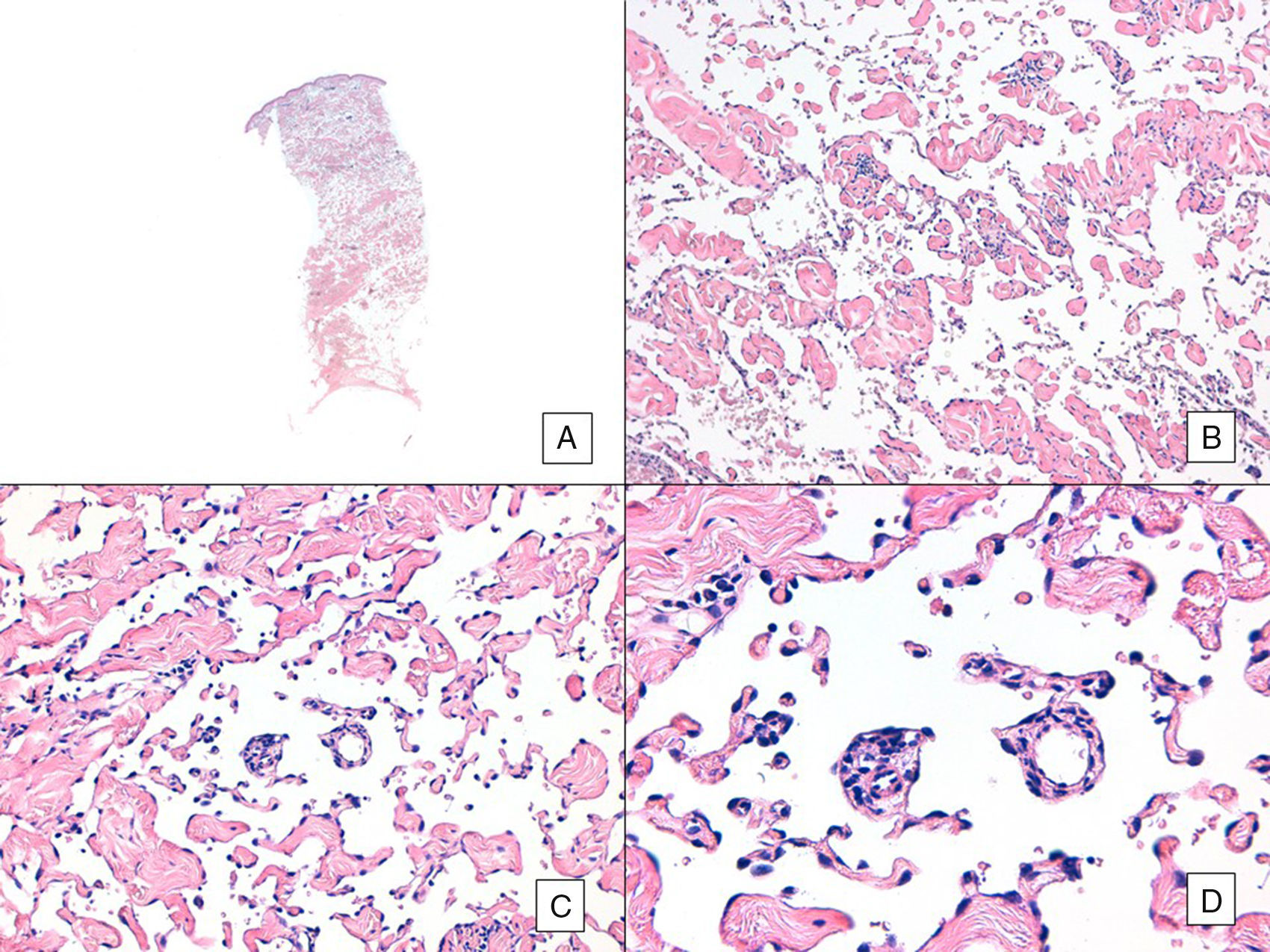
Angiosarcoma con predominio de patrón vasoformativo. A. En la imagen panorámica se aprecia infiltración de toda la dermis hasta la hipodermis (hematoxilina-eosina ×10). B. Las luces vasculares neoplásicas disecan el colágeno masivamente(hematoxilina-eosina ×100). C y D. Los vasos no neoplásicos son disecados por los endotelios neoplásicos, de modo que quedan «flotando» en la dermis (signo del promontorio) (hematoxilina-eosina ×200 y ×400, respectivamente).
Otros estudios proponen como factores histológicos de alto riesgo la presencia de necrosis, la morfología epitelioide de las células y la mayor profundidad de la invasión5,7. Nuestros resultados van en línea con lo descrito en este sentido, ya que los 3 parámetros fueron más comunes en el grupo de pacientes fallecidos. Así, encontramos con más frecuencia necrosis en el grupo de los fallecidos (5/10) que en el grupo de los vivos (1/6). Igualmente, los únicos 2 AS en los que el tipo celular predominante fue el fusocelular permanecían vivos al final de este estudio, mientras que en el grupo de los fallecidos no hubo ningún caso con predominio fusocelular. Por último, los únicos 2 casos que al diagnóstico infiltraban un plano más profundo (el músculo) pertenecieron al grupo de los fallecidos. Con respecto a las mitosis el número medio de mitosis fue superior en el grupo de mala evolución (18,3 mitosis) con respecto al de pacientes vivos (10 mitosis).
El tratamiento de elección del AS cutáneo es la cirugía con amplios márgenes seguida de radioterapia6. El frecuente carácter multicéntrico del AS, así como la habitual extensión tumoral más allá de los márgenes clínicamente apreciables, dificulta en muchos casos obtener márgenes adecuados. Además, no hay consenso en relación con los centímetros de margen quirúrgico recomendables, de modo que en la mayoría de los trabajos se dan indicaciones imprecisas como extirpación con «márgenes amplios». En nuestros casos la cirugía fue el tratamiento de referencia y se empleó en 14 de los 16 pacientes. En los casos en los que fue técnicamente factible se realizó extirpación con 3cm de margen, o en su defecto con 2cm con respecto al límite tumoral clínicamente apreciable. Estos 14 pacientes fueron sometidos a cirugía radical de su AS, mientras que los 2 casos restantes no fueron quirúrgicos de entrada, y ambos recibieron tratamiento paliativo con quimioterapia. Estos únicos 2 casos inoperables fallecieron durante el seguimiento. La radioterapia solo se empleó en 4 pacientes, y siempre de forma adyuvante a la cirugía sobre el lecho quirúrgico. Probablemente el reducido empleo de la radioterapia en el manejo de los AS de nuestra serie se pueda explicar por la gran cantidad de casos correspondientes a AS radioinducidos, que conlleva una cierta resistencia al empleo de esta arma terapéutica en tales casos. Sin embargo, en la literatura está aceptado el empleo de la radioterapia en casos de AS radioinducidos, incluso en ocasiones como tratamiento exclusivo sin cirugía23,24. La quimioterapia se empleó en la mitad de nuestros pacientes con un papel meramente paliativo y pobres resultados, como ya hemos comentado. Aunque no fue el más utilizado en nuestra serie por ser muchos casos antiguos, actualmente se suele emplear el paclitaxel en monoterapia como primera opción de la quimioterapia. Pese a las expectativas generadas, los resultados obtenidos con los fármacos angiogénicos (sunitinib, sorafenib, bavacizumab, talidomida) son decepcionantes y no se emplean.
Pese al reducido tamaño de nuestra serie, que nos ha impedido hacer un análisis estadístico de nuestros resultados, hemos observado que el mayor tamaño y la edad más avanzada se han asociado a peor pronóstico en nuestros AS. También, aunque de forma menos llamativa, se han asociado a peor pronóstico en nuestros casos los siguientes rasgos histológicos: la presencia de necrosis, el predominio celular epitelioide, la infiltración a planos más profundos y un mayor número de mitosis.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


