






Primary cutaneous angiosarcoma is one of the most aggressive skin tumors and carries a very poor prognosis. Its initially indolent clinical presentation explains the frequently late diagnosis that, together with its typically multifocal pattern and poor delimitation, often makes surgery difficult. The low incidence of primary cutaneous angiosarcoma means that few large single-center series have been published. We review the clinical and pathologic characteristics of cutaneous angiosarcomas treated in our hospital, looking for prognostic factors and for possible diagnostic traits that could facilitate early diagnosis.
Material and methodsThis was a retrospective observational study including all patients diagnosed with cutaneous angiosarcoma in Instituto Valenciano de Oncología in Valencia, Spain between January 2000 and December 2015. We recorded 16 clinical parameters, including age, sex, type of angiosarcoma, site, size, and time since diagnosis, and 8 histopathologic parameters.
ResultsWe identified 16 patients (11 women and 5 men) with cutaneous angiosarcoma. Their mean age was 67 years (median, 71 years). The most common site was the trunk (10 cases), followed by the head and neck (5 cases). The mean size of the tumor was 10cm (median, 6.5cm). Fourteen patients underwent surgical excision. Six of the 16 patients were alive at the end of the study, after a mean follow-up period of 42.5 months.
ConclusionsThe major determinants of survival among patients with cutaneous angiosarcoma are tumor size and patient age. Other characteristics associated with a poor prognosis were infiltration of deep planes (muscle), a predominantly solid histologic pattern, and a larger number of mitoses.
El angiosarcoma primario cutáneo es uno de los tumores más agresivos y de peor pronóstico de la piel. Su clínica inicialmente indolente justifica frecuentes diagnósticos tardíos, lo que sumado a su carácter muchas veces multifocal y a su mala delimitación suele dificultar la cirugía. Debido a su baja frecuencia existen pocas series largas de casos tratados en un mismo centro. Revisamos las características clínico-patológicas de los angiosarcomas cutáneos tratados en nuestro centro en búsqueda de factores pronósticos, así como de posibles rasgos que faciliten un diagnóstico precoz.
Material y métodosSe realizó un estudio observacional retrospectivo de todos los pacientes diagnosticados de angiosarcoma cutáneo atendidos en nuestro hospital entre enero de 2000 y diciembre de 2015. Se recogieron 16 parámetros clínicos incluidos —entre otros—edad, sexo, tipo de angiosarcoma, localización, tamaño, tiempo de evolución y además 8 parámetros histopatológicos.
ResultadosSe recogieron 16 pacientes con angiosarcoma cutáneo —11 mujeres y 5 varones—, la media de edad fue de 67 años y la mediana de 71 años. La localización más frecuente fue el tronco con 10 casos, seguida de la cabeza y el cuello con 5 casos. La media del tamaño tumoral fue de 10cm y la mediana de 6,5cm. Se realizó escisión quirúrgica del tumor a 14 pacientes. Tras una media de seguimiento de 42,5 meses, 6 de los 16 pacientes seguían vivos al finalizar el estudio.
ConclusionesLa supervivencia de los pacientes con angiosarcoma cutáneo viene determinada principalmente por el tamaño tumoral y la edad. Otros rasgos asociados a peor pronóstico en nuestros pacientes fueron la infiltración a planos más profundos (músculo), un patrón histológico predominantemente sólido y un mayor número de mitosis.
Cutaneous angiosarcoma has one of the worst prognoses among all cutaneous tumors. It is very aggressive and has high local recurrence rates. The 5-year survival rate is between 12% and 34% according to most studies,1,2 but it can be as high as 62%.3 Unlike other sarcomas, degree of differentiation has not been associated with prognosis in cutaneous angiosarcoma.4
The classic form of cutaneous angiosarcoma is an ill-defined bruise-like edematous lesion with a largely indolent clinical presentation in its early phases. It occurs on the face or scalp of elderly patients (Wilson-Jones angiosarcoma) and accounts for approximately 50% of all primary cutaneous angiosarcomas.5–8 Two other typical forms of angiosarcoma are Stewart-Treves syndrome, which develops in areas of long-standing lymphedema and is particularly common in women who have undergone radical mastectomy,9–11and postradiation angiosarcoma, which develops in areas of irradiated skin, particularly in the pectoral region of women with a history of breast cancer treated with radiation therapy.12–15
The histopathologic appearance of cutaneous angiosarcoma varies from relatively differentiated forms with recognizable vascular spaces covered by prominent endothelial cells with some atypia and an infiltrative pattern dissecting the collagen bundles to more solid highly undifferentiated forms composed of spindle or epithelioid cells with considerably greater atypia and pleomorphism and a higher number of mitoses. Vascular spaces are rare and the tumors can sometimes mimic carcinoma.
The mainstay treatment for cutaneous angiosarcoma—and the only one that is potentially curative if disease-free margins are achieved—is surgical excision with wide margins followed by local radiation therapy and even, in the opinion of some authors, radiation of regional lymph nodes.6 In most cases, however, it is not easy to achieve disease-free margins due to extensive subclinical spread. In addition, these tumors are frequently multifocal. Chemotherapy has a purely palliative role in the management of cutaneous angiosarcoma.
Although cutaneous angiosarcoma is rare (accounting for less than 1% of all sarcomas), most cases of angiosarcoma originate in the skin. Due to their low prevalence, cutaneous angiosarcomas are included in series of visceral or bone angiosarcoma, which have an even more dismal prognosis.1 There are thus few large series of cutaneous angiosarcoma in the literature due to the shortage of long-term, uniform cases.2,4,5,7,16 Management of angiosarcoma is additionally often disheartening, above all in cases of advanced disease, which have a very poor prognosis despite the use of aggressive treatment from the outset. Motivated thus by the difficulties associated with the management of cutaneous angiosarcoma and the little literature available, we investigated all cases of cutaneous angiosarcoma treated at Instituto Valenciano de Oncología (IVO), in Valencia, Spain, with the aim of identifying clinical, histologic, and treatment-related factors possibly associated with prognosis. To do this, we reviewed medical records and clinical findings in search of exploratory data that could serve as a guide for early diagnosis, as patients with early disease and small tumors have a considerably improved likelihood of survival.
Material and MethodsWe conducted a retrospective observational study of all cases of cutaneous angiosarcoma treated at IVO between January 2000 and December 2015. All the information compiled was extracted from the patients’ medical records, the biopsy archive of the pathology department, and the photographic archive of our department. Of the 20 cases initially identified, 4 had to be excluded: 1 because there was no follow-up, another because there was insufficient material to determine whether the tumor was a hemangioendothelioma or an angiosarcoma, and 2 because the tumors were not primary angiosarcomas. These 2 tumors had initially been labeled as cutaneous angiosarcoma because all the histology slides showed cutaneous involvement of the breast. However, on revising the blocks, we found that the cutaneous involvement was secondary in both cases, and that the primary tumor was located in the mammary parenchyma from where it extended up to the overlying skin.
The inclusion criteria for the study were clinical findings suggestive of cutaneous angiosarcoma and histologic confirmation of the diagnosis with hematoxylin-eosin staining of biopsy specimens, supported by immunohistochemical studies, which in most cases involved CD31, CD34, D240, and Ki-67 staining.
The following variables were studied for each patient: age, sex, tumor location and size, type of angiosarcoma (primary, postradiation, lymphedema-associated), treatment (surgery, radiation therapy, chemotherapy), recurrence, metastasis, survival, and death. For postradiation and lymphedema-associated tumors, we also recorded the type of previous tumor and the number of years since radiation therapy or lymphedema. The histologic variables analyzed were margin status, histopathologic pattern (vasoformative, solid, or mixed), predominant cell type (epithelioid or spindle cell), presence of necrosis (yes, no), level of invasion (epidermis, dermis, hypodermis, muscle, bone), lymphocytic reaction, infiltrative pattern, and number of mitoses per 10 fields.
ResultsSixteen cases of cutaneous angiosarcoma were included in the study. They corresponded to 11 women and 5 men aged between 35 and 83 years (mean, 67 years; median, 71 years). Ten of the cases were postradiation angiosarcoma (10 cases), 5 were Wilson-Jones angiosarcoma, and just 1 was lymphedema-associated angiosarcoma. The most common location was the trunk (10 cases), followed by the head and neck (5 cases). The upper extremities were involved in just 1 case. The smallest tumor size was 1cm and the largest was 50cm (mean, 10cm; median, 6.5cm).
Eleven of the patients had a history of cancer (breast cancer in 10 cases and seminoma in 1). With the exception of 1 case of invasive lobular carcinoma, all the breast cancers were invasive ductal carcinomas.
The mean time between radiation therapy and development of angiosarcoma in the 10 cases of postradiation angiosarcoma was 8.2 years. Just 1 of the cases appeared within 5 years of radiation therapy; the rest appeared at least 5 years later.
Fourteen cases were treated surgically and adjuvant radiation therapy was used in 4 of these. Eight patients received chemotherapy and this was the first and only treatment in 2 patients.
Doxorubicin and taxol were each used in 4 cases, ifosfamide was used in 3 cases, and paclitaxel and dacarbazine were used in 1 case each. Response to chemotherapy was poor, and although almost all the patients showed partial response, the disease progressed in all cases and the patients died during follow-up (8/8).
Five patients had distant metastasis, which involved multiple sites in most cases. The most common sites were the lung and the liver.
Ten of the 16 patients died of angiosarcoma during follow-up. The other 6 patients are currently free of disease. Mean follow-up duration was 42.5 months (median, 26 months; range, 7-188 months).
Histologically, 8 cases had a solid growth pattern, 4 had a vasoformative pattern, and 4 had a mixed pattern. The predominant cell type was epithelioid in 14 cases and spindle in just 2. Necrosis was observed in 6 tumors and the infiltrative pattern was subcutaneous in most cases (n=10). Four cases were confined to the dermis and just 2 affected the muscle planes. The surgical margins were not evaluable in 3 cases. Of the remaining cases, 8 had negative margins and 5 had positive margins. The lymphocytic reaction was mild or moderate in 10 cases, intense in 2, and inexistent in 4. In the 14 cases with a lymphocytic reaction, the infiltrate was peritumoral in 2 cases, intratumoral in 8, and mixed in 2. There was a mean of 15 mitoses per 10 fields (range, 0-37 mitoses).
The most relevant clinical and pathological results are summarized in Table 1. The results of the comparison between survivors and nonsurvivors are summarized in Table 2.
Selection of Clinical and Pathological Results for the 16 Cutaneous Angiosarcomas.a
| Patient | Age, y | Sex | Type | Location | Size, cm | Time Since Rx, mo | Dose, Gy | Previous Tumor | Breast Cancer Type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Left breast | 18 | 60 | 46 | Breast | IDC |
| 2 | 71 | F | ST | Left arm | 12 | – | - | Breast | IDC |
| 3 | 51 | F | PR | Right submammary region | 1 | 57 | 50 | Breast | IDC |
| 4 | 76 | F | WJ | Head and neck | 3 | – | – | No | . |
| 5 | 77 | F | PR | Right breast | 1 | 94 | 46 | Breast | IDC |
| 6 | 71 | F | PR | Left breast | 50 | 171 | 48 | Breast | IDC |
| 7 | 48 | M | PR | Abdominal wall | 2 | 96 | 26 | Seminoma | – |
| 8 | 55 | F | PR | Left breast | 10 | 88 | 46 | Breast | ILC |
| 9 | 69 | F | PR | Left breast | 8 | 143 | 46 | Breast | IDC |
| 10 | 76 | M | WJ | Right cheek | 6 | – | – | No | - |
| 11 | 35 | F | PR | Right breast | 12 | 66 | 50 | Breast | IDC |
| 12 | 57 | F | PR | Right breast | 8 | 108 | 50 | Breast | IDC |
| 13 | 68 | M | WJ | Head and neck | 2 | – | – | No | - |
| 14 | 80 | M | WJ | Head and neck | 15 | – | - | No | - |
| 15 | 79 | M | WJ | Head and neck | 2 | - | - | No | - |
| 16 | 83 | F | PR | Left breast | 3 | 110 | 50 | Breast | IDC |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 breast, 5 head and neck, 1 abdomen, 1 upper limb | X=10 | X=100.3 | X=45.8 | 10 breast, 1 seminoma | 10 breast cancers: 9 IDC, 1 ILC |
| Patient | Surgery | Margin, cm | AS Treatment | Death | HP Pattern | Cell Type | Necrosis | DoI | Mitoses/mm2 | Survival, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes | 0.2 | 13 | Yes | 1 | E | No | 3 | 7 | 24 |
| 2 | No | ns | 3 | Yes | 2 | E | No | 3 | 37 | 8 |
| 3 | Yes | 3.5 | 1 | No | 1 | E | No | 3 | 2 | 29 |
| 4 | Yes | 2 | 123 | Yes | 2 | E | No | 3 | 28 | 26 |
| 5 | Yes | ns | 1 | Yes | 2 | E | No | 2 | 14 | 8 |
| 6 | No | ns | 3 | Yes | 1 | E | No | 2 | 0 | 19 |
| 7 | Yes | ns | 1 | No | 3 | E | No | 3 | 6 | 187 |
| 8 | Yes | ns | 13 | Yes | 2 | E | Yes | 4 | 6 | 28 |
| 9 | Yes | ns | 13 | Yes | 3 | E | Yes | 3 | 22 | 24 |
| 10 | Yes | ns | 12 | Yes | 2 | E | Yes | 3 | 16 | 7 |
| 11 | Yes | 0.5 | 1 | No | 2 | SC | No | 3 | 18 | 76 |
| 12 | Yes | 0 | 13 | Yes | 3 | E | Yes | 3 | 36 | 26 |
| 13 | Yes | 2 | 12 | No | 3 | E | No | 3 | 5 | 95 |
| 14 | Yes | 2 | 123 | Yes | 2 | E | Yes | 4 | 17 | 24 |
| 15 | Yes | 2 | 1 | No | 2 | E | Yes | 2 | 6 | 53 |
| 16 | Yes | 3 | 1 | No | 1 | SC | No | 2 | 23 | 51 |
| 14 yes, 2 no | 1.68 | 14: Qx 4: Rt 8: Qt | 10 yes 6 no | 4 vasof 8 solid 4 mixed | 14 E, 2 SC | 6 Yes 10 No | 4 dermis 10 hypodermis 2 muscle | X=15 | X=42.8 |
The patients who died are shown in bold.
AS treatment, angiosarcoma treatment (1, surgery [Qx]; 2, radiation therapy [Rt]; 3, chemotherapy [Qt]); DoI, depth of invasion (2, dermis; 3, hypodermis; 4, muscle); E, epithelioid; HP, histopathologic (1, vasoformative [vasof]; 2,solid; 3, mixed); IDC, invasive ductal carcinoma; ILC, invasive lobular carcinoma; M, man; ns, not specified; PR, postradiation angiosarcoma; Rx, radiation; SC, spindle cell; ST, Stewart-Treves angiosarcoma; W, woman; WJ, Wilson-Jones angiosarcoma: X, mean.
Comparison of Variables Between Survivors and Patients Who Died of Cutaneous Angiosarcoma.
| Variable | Alive (n=6) | Deceased (n=10) |
|---|---|---|
| Age, mean, y | 61 | 71 |
| Women | 3 | 8 |
| Men | 3 | 2 |
| Postradiation | 4 | 6 |
| Idiopathic | 2 | 3 |
| Lymphedema-associated | 0 | 1 |
| Trunk | 4 | 6 |
| Head and neck | 2 | 3 |
| Upper limbs | 0 | 1 |
| Size, cm | 3.6 | 13.1 |
| Time since radiation therapy, mo | 82.25 | 110.6 |
| Radiation dose, Gy | 44 | 47 |
| Breast | 3 | 7 |
| Seminoma | 1 | |
| Surgery | 6 | 8 |
| Radiation therapy | 1 | 3 |
| Chemotherapy | 0 | 8 |
| Vasoformative | 2 | 2 |
| Solid | 2 | 6 |
| Mixed | 2 | 2 |
| Necrosis | 1 | 5 |
| Dermis | 2 | 2 |
| Subcutaneous | 4 | 6 |
| Muscle | 0 | 2 |
| Mitoses | 10 | 18.3 |
| Survival, mo | 81.8 | 19.4 |
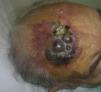
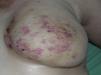
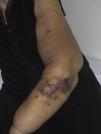
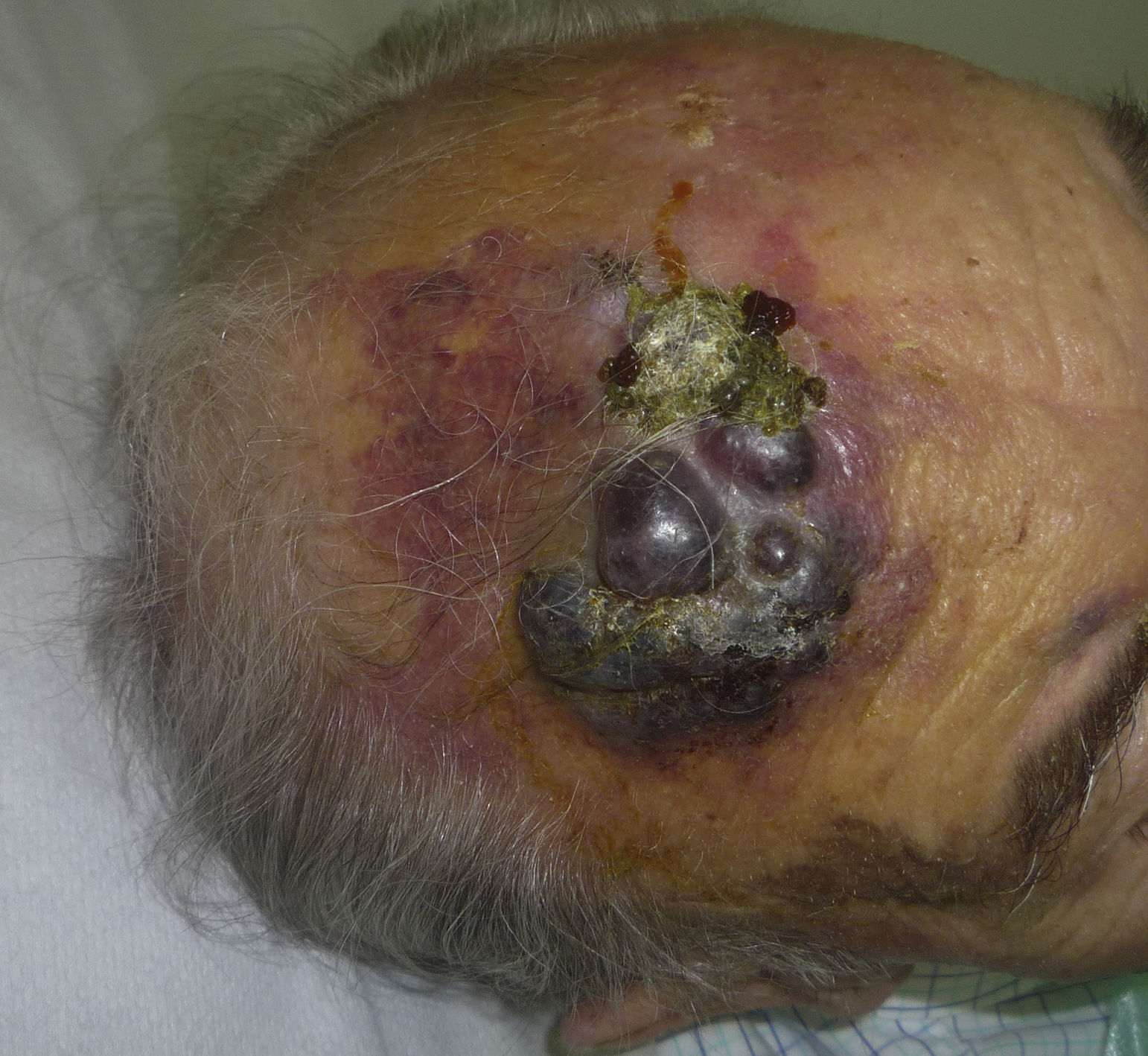
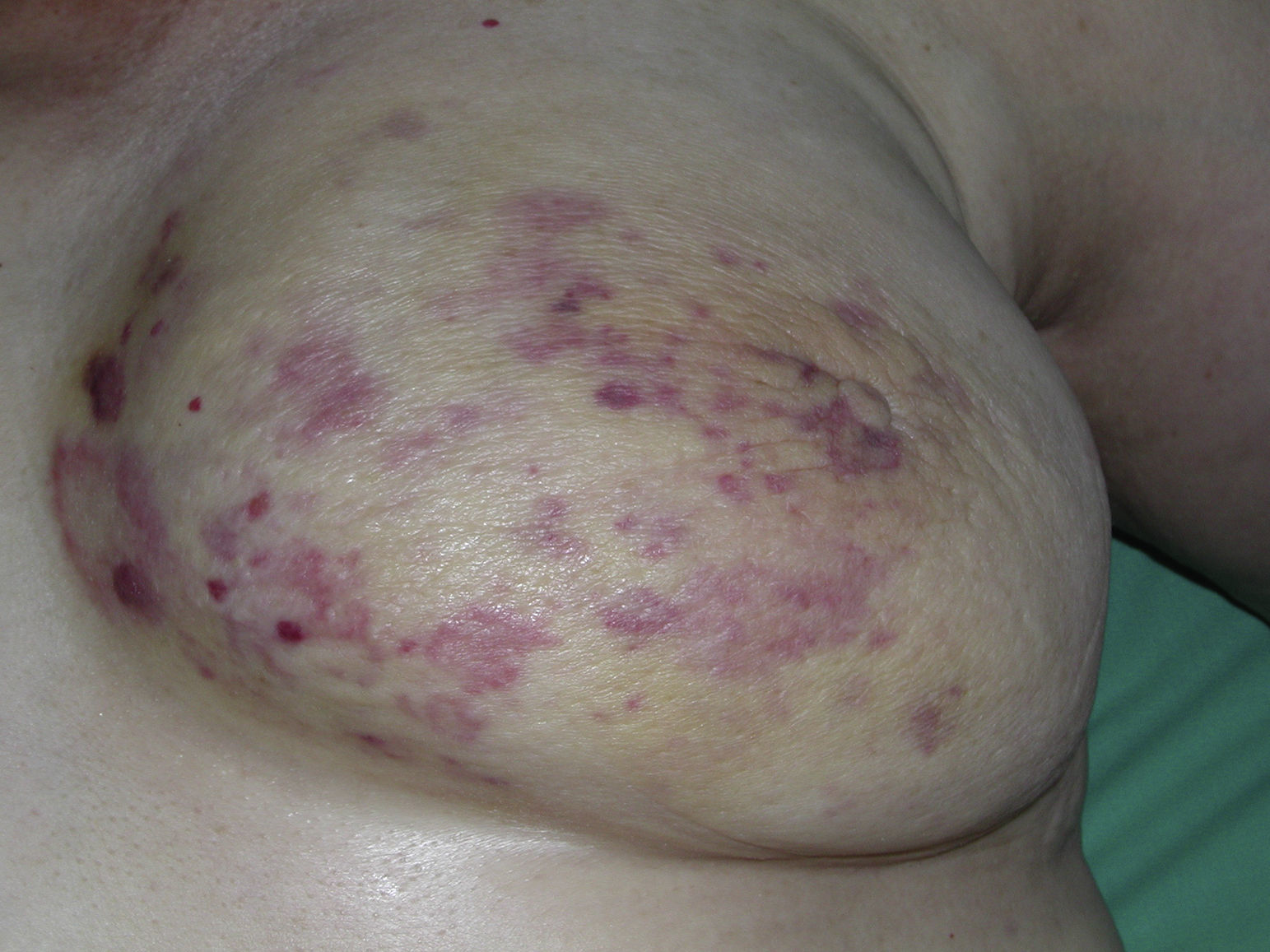
Cutaneous angiosarcoma is a very rare tumor, evidenced by the fact that we were only able to collect data on 16 tumors diagnosed over a period of 14 years at an oncology hospital. Overall, cutaneous angiosarcoma is somewhat more common in elderly men. This is because the most common form of cutaneous angiosarcoma in the general population is primary head and neck angiosarcoma (also known as idiopathic or Wilson-Jones angiosarcoma)(Fig. 1), which tends to affect elderly men.5,7,17 Postradiation angiosarcoma (Fig. 2) is now the second most common form of cutaneous angiosarcoma due to the increased use of radiation therapy as opposed to radical mastectomy to treat breast cancer.2,13,18 This change has also led to a reduction in the frequency of lymphedema-associated angiosarcoma, which is currently the least common form of this tumor. The prevalence of the different forms of cutaneous angiosarcoma in our series does not coincide with reports in the literature as our hospital treats a large number of women with breast cancer, explaining why the most common form of cutaneous angiosarcoma at our hospital was postradiation angiosarcoma. This predominance of breast cancer cases also explains why 11 of the 16 patients were women. There was just 1 case of lymphedema-associated angiosarcoma (Fig. 3), which does coincide with prevalence rates reported elsewhere. The patient had chronic lymphedema of the left arm secondary to axillary lymph node dissection performed as part of breast cancer treatment 22 years earlier.
With the exception of 1 woman who had idiopathic or Wilson-Jones angiosarcoma, the rest of the women in our series (n=10) had a history of breast cancer. Nine of these cancers were invasive ductal carcinomas. As 80% of breast cancers in the general population are invasive ductal carcinomas and just 10% are lobular carcinomas, the high prevalence of invasive ductal carcinoma in our study is probably a reflection of the high prevalence of this cancer in the general population, rather than of a particular association between postradiation cutaneous angiosarcoma and invasive ductal carcinoma, as our results would seem to suggest. In other words, the proportions observed in our series are in consonance with the rates described for different breast cancers in the general population. There were 2 cases of breast angiosarcoma initially included in our study that are worthy of mention. It has been reported that breast angiosarcoma tends to cutaneous when it is induced by radiation and parenchymal when it is not.19 In our series, of the 18 cases of cutaneous angiosarcoma initially identified, there were 2 cases of breast angiosarcoma not induced by radiation therapy. On reviewing these cases, we found that the primary location of the tumor was the mammary parenchyma and not the skin. The tumors were therefore excluded from the study as they were secondary not primary. The remaining cases were all primary breast angiosarcomas induced by radiation, in line with reports in the literature.
The most common location for angiosarcoma in our series was the breast (10 cases) and not the face or scalp, as would be expected. The only case that involved the extremities was a lymphedema-associated angiosarcoma. Again, the fact that the most common tumor site was the breast would be explained by the predominance of radiation-induced tumors in our series.
Although the latency period between exposure to radiation therapy and the development of angiosarcoma is highly variable according to reports in the literature (3-50 years), it tends to be longer (on average, 25 years) when the disease treated by radiation is benign. Latency periods reported for malignant diseases are shorter (around 10-15 years), except in the case of angiosarcoma of the breast, for which a mean of approximately 5 years has been described.12 The reason for this shorter period in the case of breast angiosarcoma is unclear, although several theories have been proposed, including the large volume of irradiated skin, the presence of associated lymphedema, factors that are possibly intrinsic to the breast, and a possible synergic effect with chemotherapy.13,14 In our 10 postradiation cases, angiosarcoma was diagnosed after a mean of 8.2 years, and at least 5 years had passed in 90% of cases. No association between latency period and prognosis has been reported for angiosarcoma. In our series, mean time from radiation therapy to development of angiosarcoma was somewhat longer in patients who died (110.6 months) than in those who survived (82.25 months). In the only case of Stewart-Treves syndrome, the patient had undergone lymph node dissection 22 years earlier. Latency periods reported for lymphedema-associated angiosarcoma are highly variable, with a range from 1 to 30 years and a mean of 10. Stewart-Treves syndrome accounts for 90% of all cases of lymphedema-associated angiosarcoma.9,11 Angiosarcoma can, however, also arise in other forms of lymphedema, such as congenital lymphedema, filarial lymphedema, and lymphedema secondary to lymph node dissection in other parts of the body.10
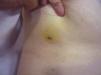
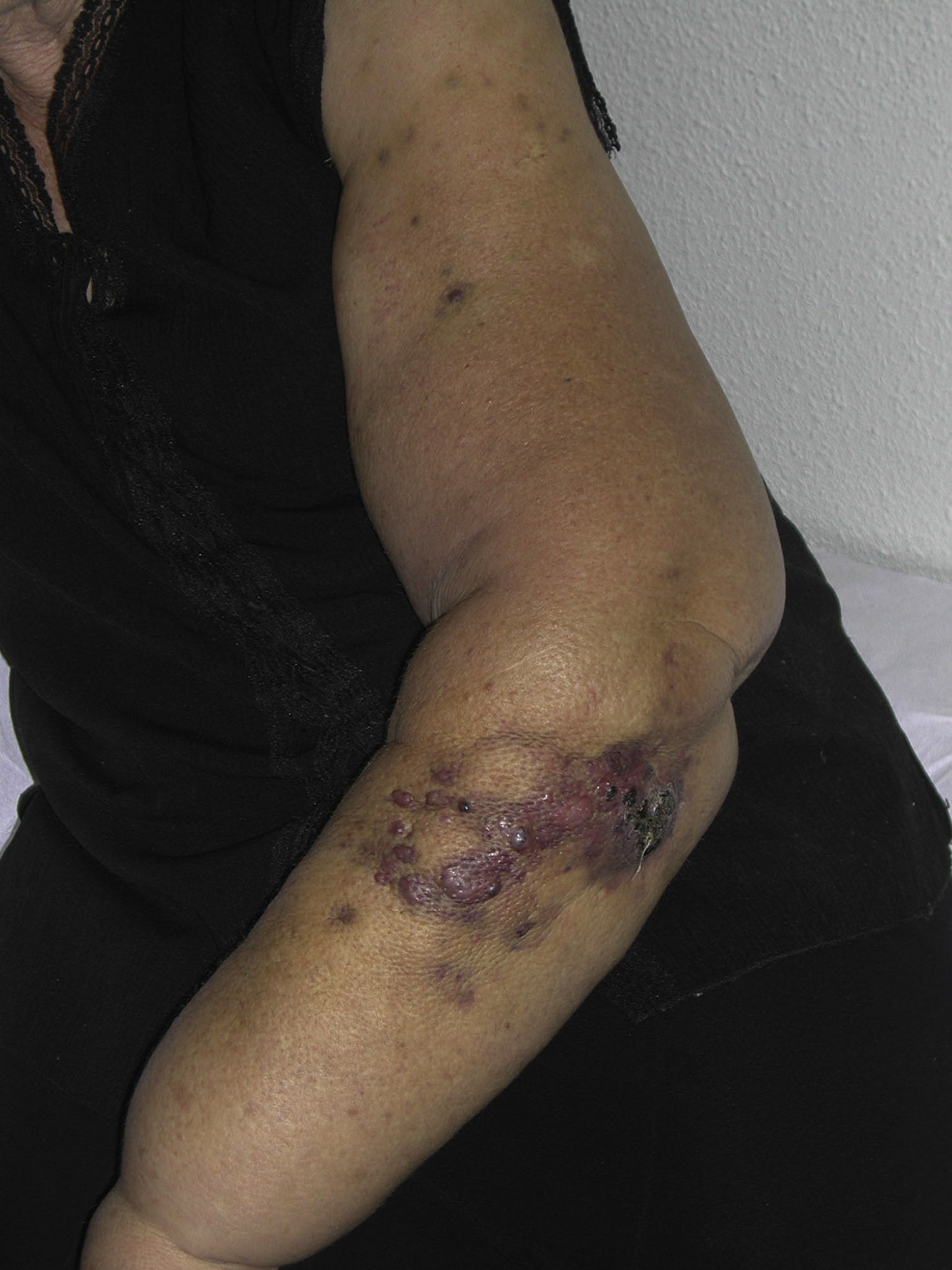
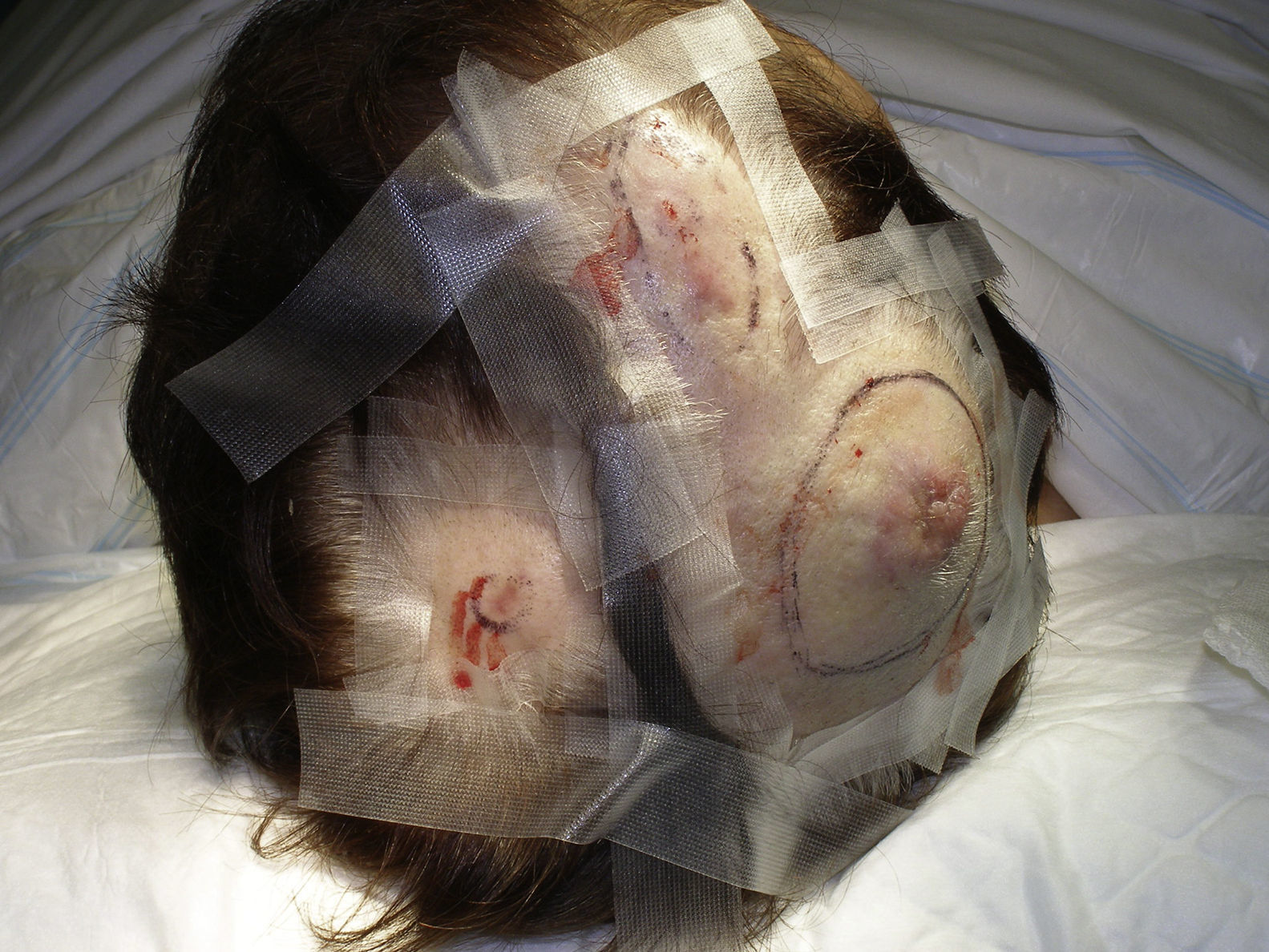
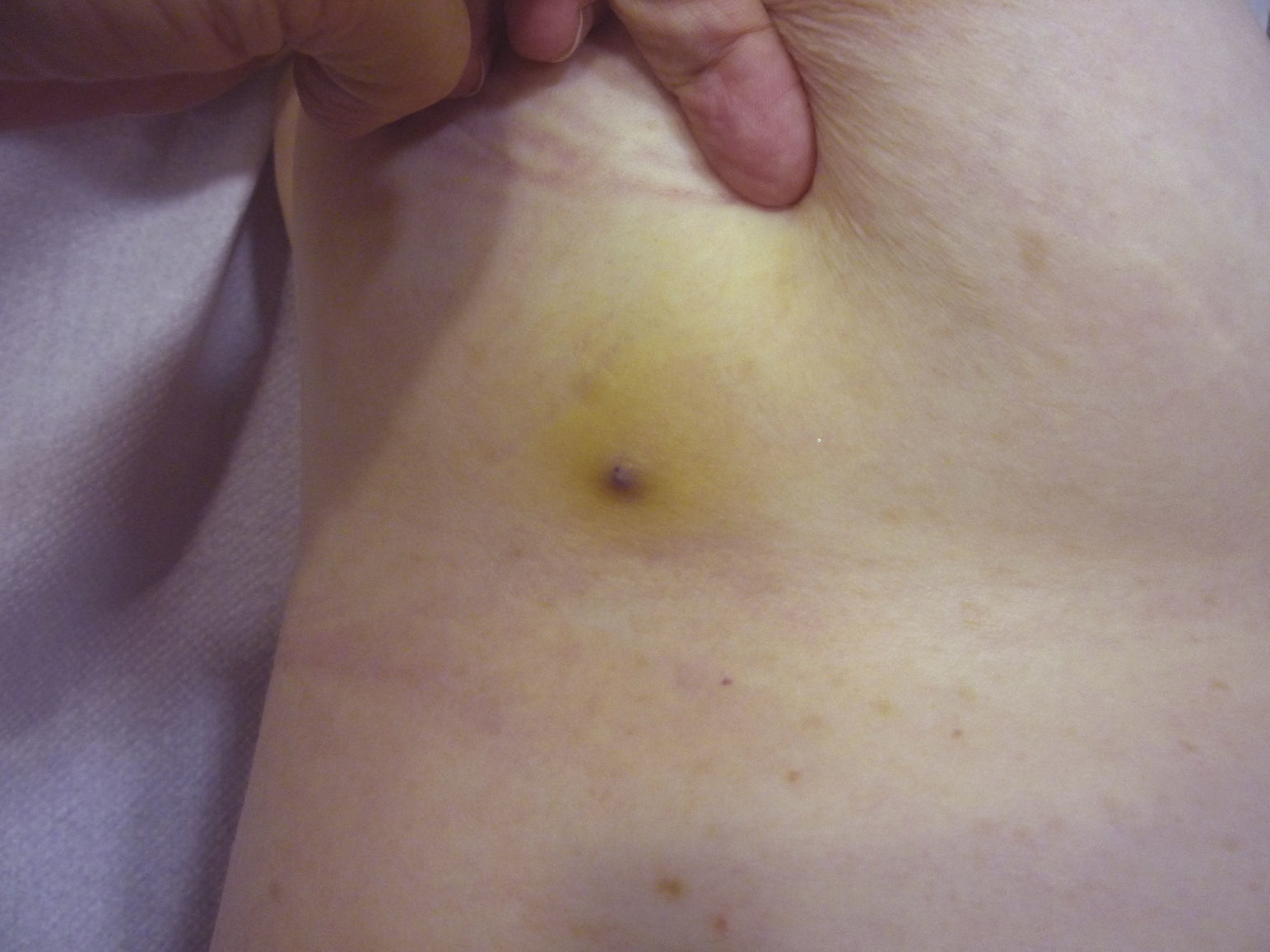
Tumor size is currently the most widely accepted prognostic marker for cutaneous angiosarcoma, and it has been frequently reported that angiosarcomas measuring 5cm or more have a worse prognosis than smaller tumors.5,17 In our series, we observed differences in mean tumor size between survivors and nonsurvivors. Those who survived had a mean tumor size of 3.6cm, which was over 3 times smaller than the mean size (13.1cm) in the group of patients who died. Our results thus support the belief that larger tumors are associated with worse outcomes in angiosarcoma,and highlight the importance of diagnosing the tumor when it starts as a bruise-like lesion, although this is particularly difficult in angiosarcomas of the scalp and even more when the patient still has hair (Fig. 4). Postradiation angiosarcoma of the breast is easier to diagnose, as any persistent lesion in this area with a vascular or bruise-like appearance should be biopsied. One potentially useful diagnostic clue is a yellowish halo (corresponding to hemosiderin) around the suspicious lesion (Fig. 5), as we have never observed this sign in benign vascular proliferations on irradiated skin. Postradiation angiosarcoma and atypical vascular proliferations on irradiated skin must be distinguished histologically but clinical features can help. Atypical vascular proliferations tend to be much smaller than angiosarcomas and they also generally have a shorter latency period.20 Unlike angiosarcomas, these proliferations are confined to the superficial and mid dermis and do not invade the subcutaneous tissue. In addition, histology does not show the characteristic nuclear atypia observed in angiosarcoma, nor the multiple layers of endothelial cells or mitotic figures. Nevertheless, it can occasionally be difficult to distinguish between the 2 entities and there have been reports of the 2 lesions coexisting in the same irradiated breast. There have even been cases of radiation-induced atypical vascular proliferations progressing to angiosarcoma.21 In very complicated cases, it can be helpful to search for overexpression of the MYC gene if an immunohistochemical study is performed, or amplification if fluorescent in situ hybridization is used. MYC amplification is quite common in secondary angiosarcomas, but does not appear to occur in atypical vascular proliferations on irradiated skin.22
The other most widely accepted prognostic marker in angiosarcoma is age. In our series, the patients who survived were on average younger than those who died (62 versus 71 years). Notwithstanding, 10 of the 16 patients in our series had died by the end of the study, corroborating the poor prognosis generally associated with angiosarcoma.
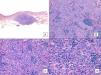
Unlike the case of other sarcomas, the American Joint Committee on Cancer does not consider histologic grade to be a relevant prognostic marker in angiosarcoma. Several authors, however, have suggested that certain histopathologic features might have a bearing on prognosis. Some recent studies, for example, have claimed that a predominant solid pattern might be a relatively good prognostic marker in angiosarcoma of the head and neck.3,16 This was not the case in our series as 6 of the 10 patients who died had this pattern compared with just 2 of the 6 patients who were still alive at the end of the study (Figs. 6 and 7).
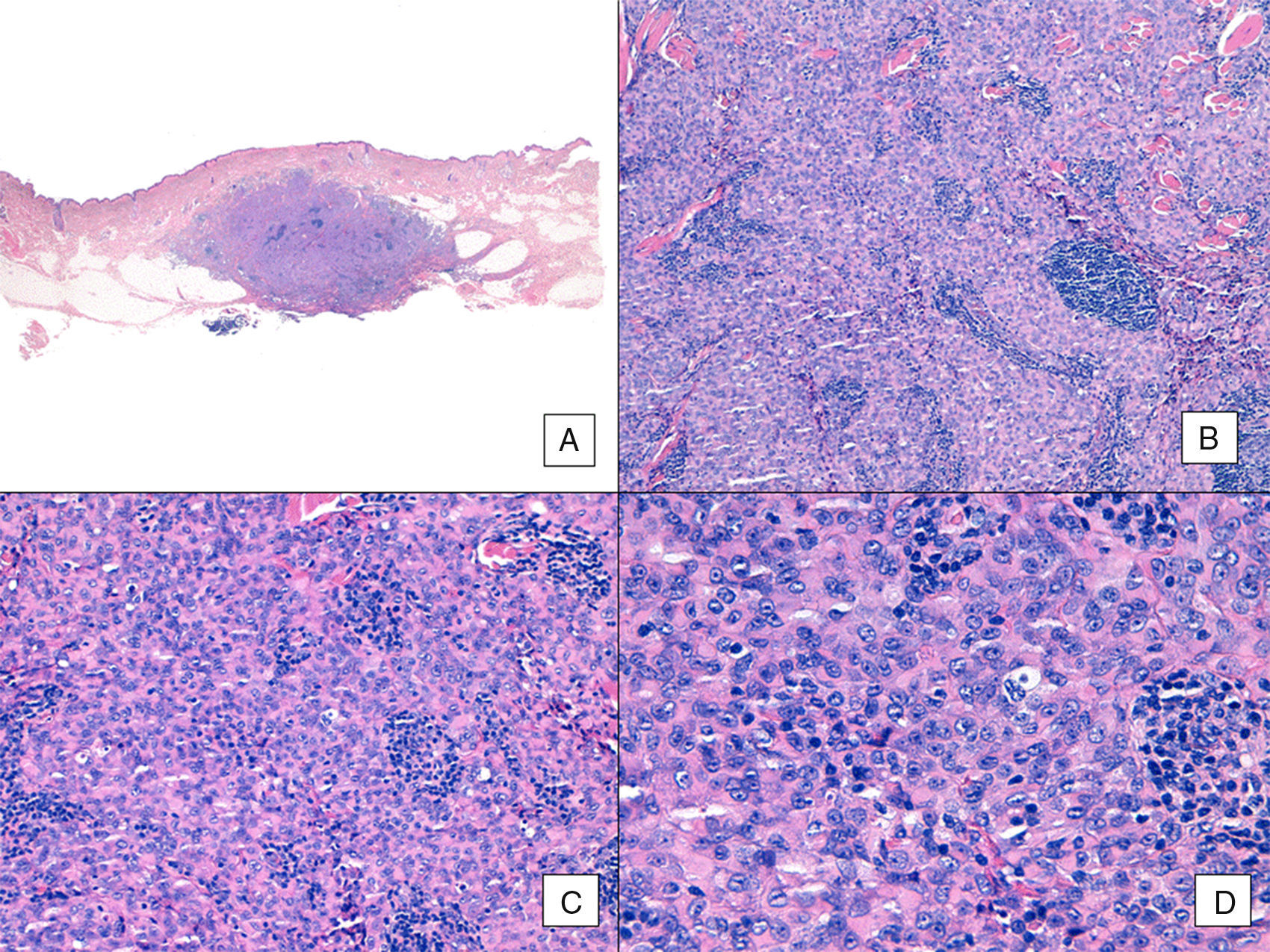
Angiosarcoma with a predominant solid pattern. A, The panoramic image show invasion of the mid and deep reticular dermis and the hypodermis (hematoxylin-eosin, original magnification ×10). B, Densely cellular tumor that has destroyed pre-existing structures and is accompanied by nodular lymphoid infiltrates (hematoxylin-eosin, original magnification ×100). C and D, Detailed view showing a predominance of epithelioid cells accompanied by a lymphocytic infiltrate (hematoxylin-eosin ×200 and ×400, respectively).
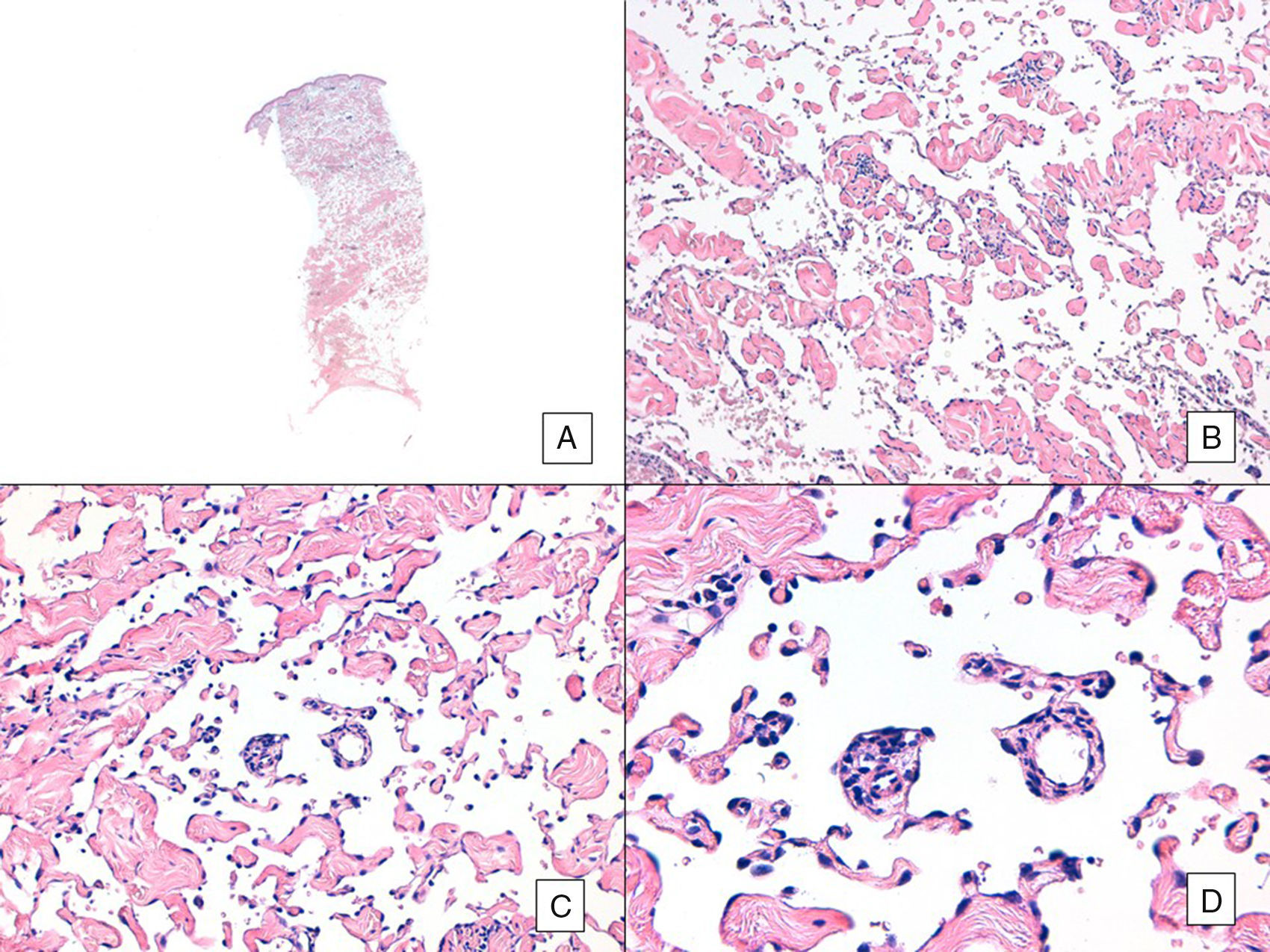
Angiosarcoma with a predominant vasoformative pattern. A, Panoramic image showing infiltration of dermis down to the hypodermis (hematoxylin-eosin, original magnification ×10). B, Neoplastic vascular spaces with pronounced collagen dissection (hematoxylin-eosin, original magnification ×100). C and D, Non-neoplastic vessels dissected by neoplastic endothelial cells, which remain “floating” in the dermis (promontory sign) (hematoxylin-eosin, original magnification ×200 and ×400, respectively).
Other potentially high-risk histologic factors proposed in the literature are presence of necrosis, an epithelioid morphology, and a greater depth of invasion.5,7 Our results support these potential markers, as all 3 variables were more common in the group of nonsurvivors. Necrosis was observed in 5 of the 10 patients who died and in just 1 of the 6 patients who survived. Likewise, the only 2 patients with a predominant spindle cell pattern were still alive at the end of the study. None of the patients who died, by contrast, had this pattern. Finally, the only 2 patients with involvement of the muscle planes died. The mean number of mitoses was also higher in the group of nonsurvivors (18.3 vs 10).
The treatment of choice of cutaneous angiosarcoma is surgical excision with wide margins followed by radiation therapy.6 The frequent multifocal nature of angiosarcoma and its propensity to develop subclinical spread often complicates the achievement of clear margins. Furthermore, there is no universal agreement on what constitutes an adequate margin, and most studies provide imprecise information, such as excision with “wide margins”. In our series, surgery was the treatment of choice in 14 of the 16 patients. Margins of 3cm were used where technically feasible, and in the other cases, clearance was attempted using 2-cm margins. The 14 patients underwent radical surgery. The other 2 were not deemed candidates for surgery and received palliative chemotherapy. They died during follow-up. Adjuvant radiation therapy to the surgical bed was used in just 4 patients. This low use of radiation therapy in the management of angiosarcoma in our series can probably be explained by the large number of cases of postradiation angiosarcomas in our series. In other words, it was probably influenced by a certain level of resistance to using radiation in these cases. Use of radiation therapy in postradiation angiosarcoma, however, finds support in the literature, and is even sometimes administered as monotherapy, without surgery.23,24 Chemotherapy was used in half of the patients. As already mentioned, it had a merely palliative role and was associated with poor outcome in all cases. Although paclitaxel monotherapy was not the most used chemotherapy regimen in our series (as the cases were diagnosed some time ago), this regimen is now used as a first-line option in most cases. Despite the initial expectations generated, angiogenic drugs (sunitinib, sorafenib, bevacizumab, thalidomide) are not used in this setting due to their disappointing results.
Despite the small number of cases in our series of cutaneous angiosarcoma, which impeded us from performing statistical analyses, we observed that larger tumor size and older age were both associated with worse prognosis. A less evident association was also observed between poor prognosis and the following histologic features: presence of necrosis, a predominance of epithelioid cells, invasion of deeper layers, and a larger number of mitoses.
Ethical DisclosuresProtection of humans and animalsThe authors declare that no tests were carried out in humans or animals for the purpose of this study.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients.
Right to privacy and informed consentThe authors declare that no private patient data appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Requena C, Sendra E, Llombart B, Sanmartín O, Guillén C, Lavernia J, et al. Angiosarcoma cutáneo: estudio clínico-patológico de 16 casos. Actas Dermosifiliogr. 2017;108:457–465.


