



INTRODUCCION
El dermatofibrosarcoma protuberans (DFSP) es un tumor poco frecuente, de malignidad intermedia, con una baja incidencia de metástasis, pero con alta frecuencia de recurrencia local. El diagnóstico histológico de este tumor en ocasiones puede resultar difícil, y es necesario en estos casos realizar un estudio inmunohistoquímico para diferenciarlo de dermatofibromas atípicos y de otros sarcomas 1,2. Sin embargo, estas técnicas no siempre son suficientes para establecer un diagnóstico adecuado. Recientemente, se han descrito en el DFSP rasgos citogenéticos específicos como la translocación recíproca t(17;22)(q22; q13) o, más a menudo, cromosomas supernumerarios en anillo derivados de la t(17;22) 3. Como consecuencia de esta translocación, se produce una fusión génica entre el gen del colágeno tipo Iα (COL1A1), en el cromosoma 17q, con el gen de la cadena β del factor de crecimiento derivado de las plaquetas (PDGFB), en el cromosoma 22q, y cuya expresión puede detectarse mediante técnicas de biología molecular, como amplificación mediante transcripción inversa y reacción en cadena de la polimerasa (RT-PCR) utilizando iniciadores específicos 4. Esta translocación es exclusiva del DFSP y del fibroblastoma de células gigantes (variedad infantil del DFSP).
En este estudio, presentamos el caso de un DFSP con componente de fibrosarcoma, donde el estudio de biología molecular realizado sobre material incluido en parafina mediante RT-PCR muestra la presencia de un transcrito de fusión que implica al exón 19 del COL1A1 y al exón 2 del PDGFB, lo que apoya el diagnóstico de DFSP.
DESCRIPCION DEL CASO
Una mujer de 37 años de edad, sin antecedentes personales de interés, fue remitida al Servicio de Dermatología de la Fundación Instituto Valenciano de Oncología para tratamiento quirúrgico de una tumoración localizada en la parte proximal del brazo izquierdo, en la región deltoidea. La paciente refería hacía 9 años la aparición de una lesión nodular de pequeño tamaño, de crecimiento lento y progresivo, extirpada en el servicio de cirugía con diagnóstico histológico de dermatofibroma. A los 6 años de la intervención apareció en la zona de la cicatriz, un nódulo subcutáneo que fue interpretado como queloide, tanto clínica como histológicamente. La paciente había sido tratada posteriormente con infiltraciones de corticoides intralesionales e imiquimod, con escasa respuesta. En los últimos 4 meses se habría producido un rápido crecimiento de la lesión, motivo por el que consultó al dermatólogo.
A la exploración física se apreciaba una tumoración sobreelevada, intradérmica de 8 × 4 cm de tamaño, muy indurada e infiltrada a la palpación (fig. 1A). La zona superficial presentaba capilares ramificados y dilatados con zonas blanquecinas, nacaradas y brillantes de aspecto cicatrizal. La resonancia magnética de la zona afectada efectuada para valorar la extensión del tumor, reveló un desplazamiento del músculo deltoides y la existencia de infiltración muscular por la lesión en dicho músculo (fig. 1B). La biopsia cutánea mostró la presencia de una tumoración en la dermis con infiltración del tejido celular subcutáneo, constituida por células fusiformes de núcleo elongado con un patrón estoriforme y otras células más pleomórficas con un mayor índice mitótico, con una disposición en largos fascículos, adoptando un patrón en espina de pescado (fig. 2A). El estudio inmunohistoquímico mostró una tinción intensa citoplasmática con CD34 en más del 90 % de las células fusiformes con patrón estoriforme, con mínima inmunotinción en las células pleomórficas de la zona de patrón en espina de pescado (fig. 2B). El factor XIIIa fue negativo en la población tumoral.
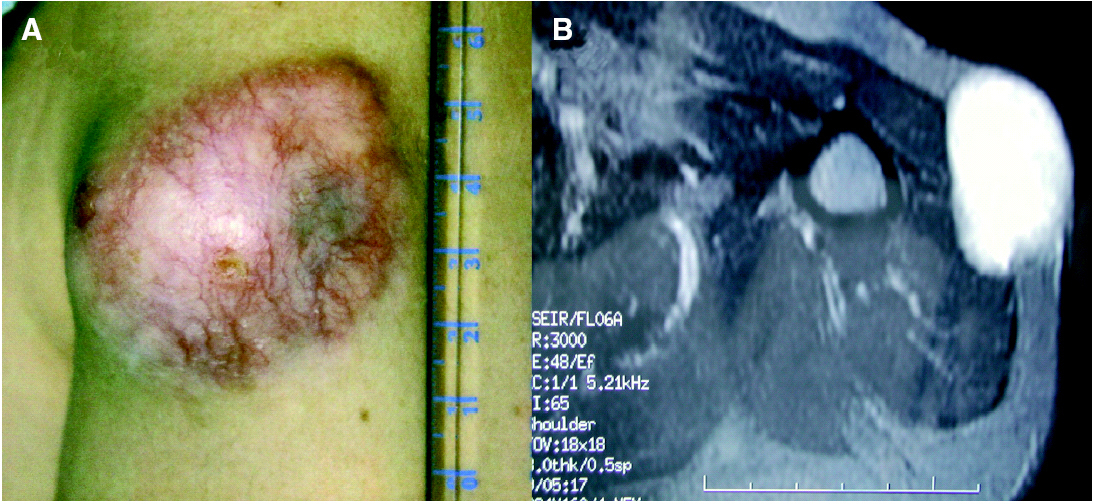
Fig. 1.--A) Tumoración localizada en la parte proximal del brazo de 8 × 4 cm. B) Resonancia magnética nuclear: infiltración por la tumoración del músculo deltoides.
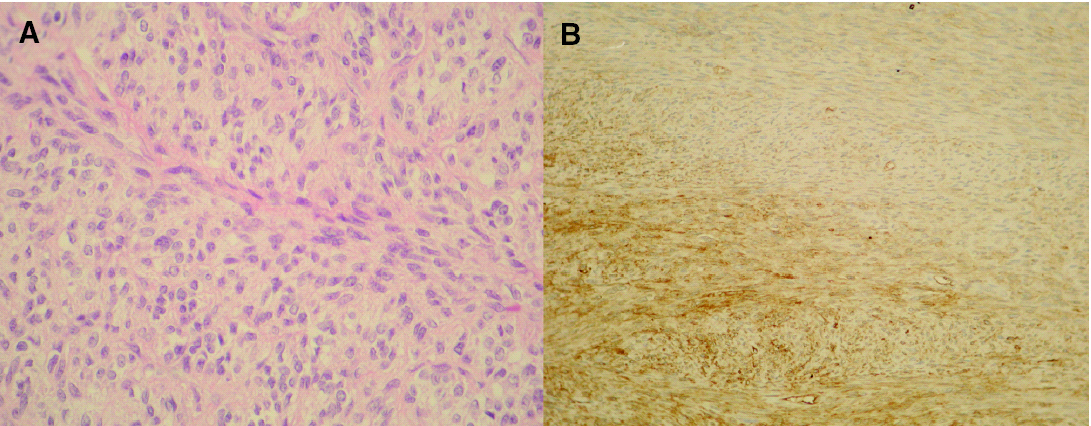
Fig. 2.--A) Células pleomórficas, con disposición en largos fascículos típicos de fibrosarcoma. (Hematoxilina-eosina, ×200.) B) Tinción intensa citoplasmática con CD34 en la zona típica de DFSP, siendo mínima la inmunotinción en las células pleomórficas de la zona de fibrosarcoma.
La paciente fue intervenida mediante cirugía micrográfica de Mohs con bloque en parafina, con exéresis en bloque del tumor, incluyendo fascículos superficiales del músculo deltoides, necesitando un estadio de 1 cm para obtener márgenes negativos. El defecto resultante, de 9 × 5 cm, se cerró mediante sutura directa. Tras un año de seguimiento, la paciente no presenta evidencia clínica ni radiológica de enfermedad.
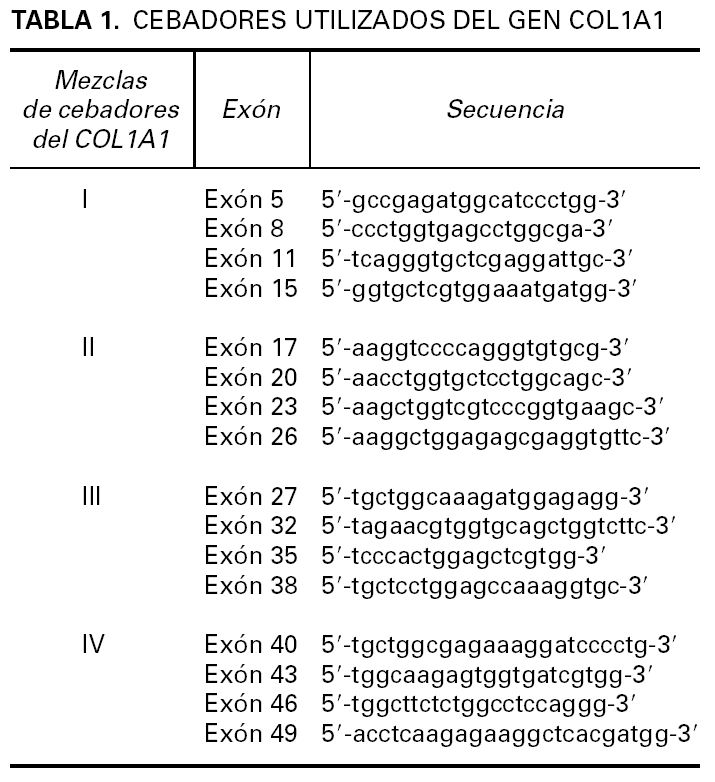
Para el estudio de biología molecular se partió de 5 cortes de 5 µm que se sometieron a un pretratamiento de desparafinación con sucesivos lavados en xilol y etanol, seguido de una digestión enzimática con proteinasa K a 55 °C durante 3 h. A continuación, se llevó a acabo la extracción de ARN siguiendo un procedimiento modificado del desarrollado por Chomzynski y Sacchi 5, que emplea el TRIZOL®Reagent (GIBCO BRL). Tras la obtención del ARN se comprobó la integridad del mismo en una electroforesis en gel de agarosa al 1 % en condiciones no desnaturalizantes en los que se correrán 1 μl de ARN. La presencia de 2 bandas, correspondientes al ARN ribosómico 28S y 18S tras tinción del gel en bromuro de etidio y visualización con luz ultravioleta, confirmaría la integridad del ARN obtenido.
Para la transcripción inversa (RT) se empleó un kit comercial y se siguieron las recomendaciones del fabricante (GeneAmp RNA PCR Core Kit, Applied Biosytems). En nuestro caso se preparó un volumen de ADNc suficiente (60 µl) como para poder realizar un cribado completo de los exones del COL1A1. En primer lugar realizamos una primera aproximación mediante 4 PCR múltiples que incluyen además de un iniciador localizado en el exón 2 del gen PDGFB (59 -atcaaaggagcggatcgagtggtc-39) (común en todas las PCR múltiples) y otros localizados a lo largo del gen COL1A1 (tabla 1) 4. También se amplificó una región del gen CDK4 como control de la presencia de ARN en la muestra analizada. El estudio mostró positividad del gen control además de en la PCR múltiple II. A continuación se volvió a amplificar el producto de ADNc con los iniciadores del Set II pero esta vez por separado confirmándose la presencia del gen de fusión COLIA1-PDGFB con un iniciador localizado en el exón 17 del gen COL1A1 y otro en el exón 2 del gen PDGFB (fig. 3A). Por último, el producto obtenido se estudió mediante secuenciación empleando el kit de secuenciación BigDye v3.1. La reacción de secuenciación se determinó mediante electroforesis capilar en un secuenciador automático ABI3130 confirmándose la fusión del exón 19 del COL1A1 con el exón 2 del PDGFB (fig. 3B).
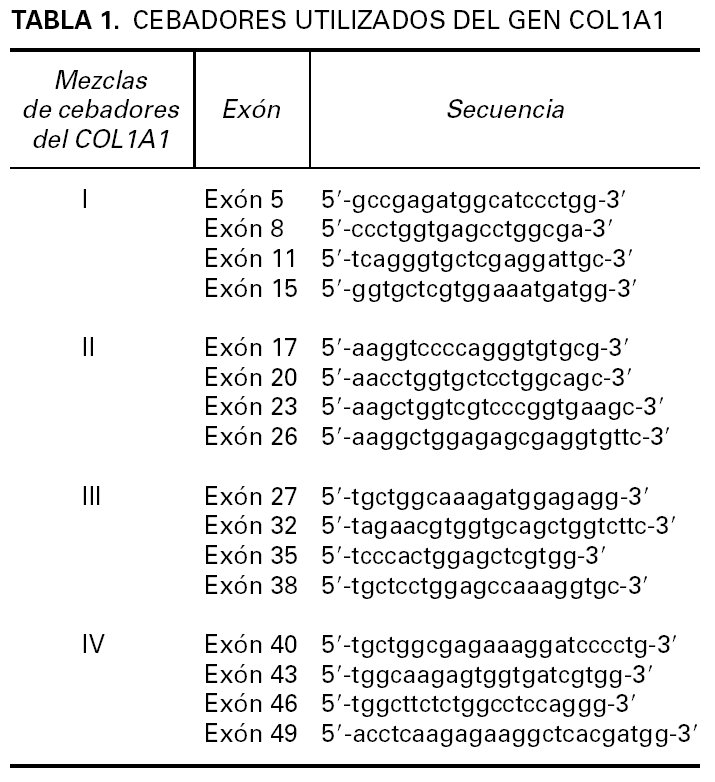
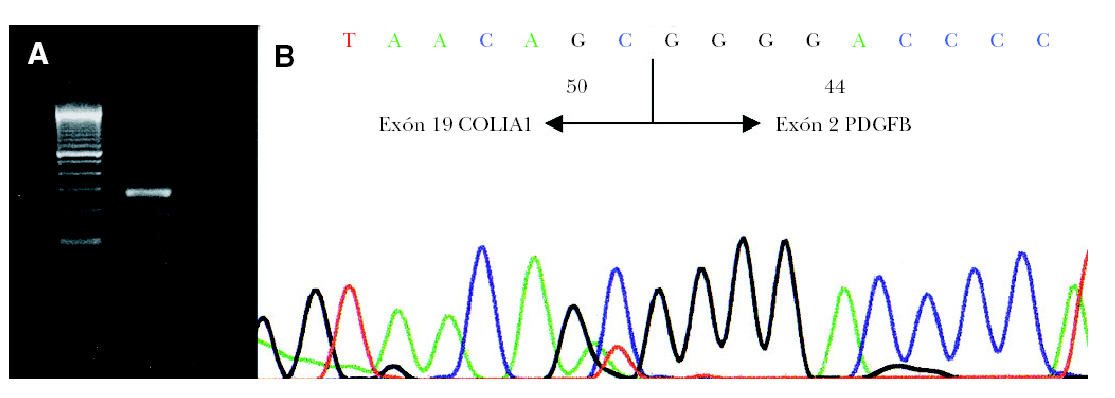
Fig. 3.--A) Detección por RT-PCR de la translocación COL1A1-PDGFB. B) Análisis de la secuenciación de nucleótidos mostrando la fusión del exón 19 COL1A1 con el exón 2 PDGFB.
DISCUSION
El DFSP es el tumor fibrohistiocitario de malignidad intermedia más frecuente, que constituye aproximadamente el 1,8 % de todos los sarcomas de partes blandas y el 0,1 % de todos los cánceres. Su incidencia se ha estimado entre 0,8 y 5 casos por millón de habitantes y año 1,2,6. Se caracteriza por un crecimiento infiltrativo lento, con poca tendencia a las metástasis a distancia, pero con una elevada capacidad de destrucción local y alta frecuencia de recidiva tras el tratamiento quirúrgico.
Histológicamente, el DFSP es un tumor intradérmico de gran tamaño, constituido por células fusiformes monomorfas de núcleo elongado con depósito de colágeno intercelular y presencia de pequeños capilares. La epidermis suele estar respetada, presentando la dermis papilar una zona libre de lesión o zona «Grenz». Las células por lo general se entrecruzan y adquieren un patrón estoriforme, con infiltración del tejido celular subcutáneo, creando una imagen en panal característico 7. Existen variedades histológicas de DFSP como la presencia de melanina en las células dendríticas (tumor de Bednar), cambios mixoides, mioides, células multinucleadas gigantes con pseudoespacios vasculares (fibroblastoma de células gigantes), y formas más pleomórficas (fibrosarcoma). En series largas descritas en la literatura médica entre el 10 y el 15 % de los casos presentan áreas de fibrosarcoma, considerándose estas lesiones de mayor agresividad biológica y de mayor riesgo metastásico 8.
Las técnicas inmunohistoquímicas son de gran utilidad en el diagnóstico diferencial con los dermatofibromas atípicos y otros sarcomas, pues hasta el 90 % de los DFSP son CD34 positivos, y únicamente el 10 % son factor XIIIa positivos 9,10. Sin embargo, en ocasiones, estas técnicas no son suficientes para establecer un diagnóstico adecuado.
La primera evidencia del gen de fusión entre los genes COL1A1 y PDGFB como consecuencia de la translocación t(17;22) con técnicas de RT-PCR en DFSP se publicó en 1995 por Minoletti et al 11. En estudios más recientes se ha demostrado que alrededor del 90 % de los casos de DFSP presentan la translocación t(17;22) 4. Sin embargo, se han tratado siempre de series cortas de pacientes, por lo que la frecuencia real de la translocación está todavía por demostrar. La región de rotura en la t(17;22) afecta en todos los casos descritos hasta la fecha al exón 2 del gen PDGFB, localizado en el cromosoma 22, que se fusiona con alguno de los exones del gen COL1A1 localizado en el cromosoma 17. Los puntos de fusión en COL1A1 son muy variables y pueden afectar a cualquiera de sus 51 exones. En nuestro caso, el punto de fusión se produce entre el exón 19 del COL1A1 y exón 2 del PDGFB y no ha sido descrito previamente. Hasta la fecha, no se ha encontrado relación entre los diferentes puntos de fusión descritos en la literatura médica con el subtipo histológico, localización anatómica, u otro factor estudiado, incluidos los parámetros de recurrencia y progresión.
La presencia del gen de fusión COL1A1-PDGFB en el DFSP puede demostrarse aislando ARN del tumor, bien de muestras fijadas e incluidas en parafina o a partir del tejido criopreservado, y utilizando procedimientos de biología molecular como el de la RT-PCR. Estas técnicas son altamente sensibles y específicas pero presentan una serie de dificultades inherentes, especialmente cuando sólo se dispone de material incluido en parafina, pues el ARN extraído en muchas ocasiones es escaso y de mala calidad lo que disminuye considerablemente la sensibilidad del procedimiento. Otra dificultad añadida en el caso de los DFSP consiste en la gran variabilidad del punto de fusión en el COL1A1, que como hemos indicado anteriormente, puede encontrarse en cualquiera de sus 51 exones. Esta heterogeneidad obliga a la optimización del proceso de detección del gen de fusión mediante PCR múltiples que abarquen la totalidad de los exones del gen COL1A1. Pero a pesar de ello, no deja de ser una técnica muy laboriosa y de resultados no siempre satisfactorios.
La proteína de fusión generada por el gen de fusión COL1A1-PDGFB, es procesada a nivel extracelular hasta transformarse en PDGFB completamente maduro y funcional, capaz de inducir un potente estímulo mitógeno mediante la activación de su receptor 12. Por tanto, la consecuencia fundamental de la translocación t(17;22) es inducir una activación del receptor del PDGFB, mediante la producción autocrina y paracrina de su ligando funcional.
El conocimiento de este mecanismo de acción del producto de fusión COL1A1-PDGFB en DFSP ha sugerido que el empleo de inhibidores de la familia de las tirosincinasa, como el mesilato de imatinib puede ser útil en casos de DFSP no susceptibles de tratamiento quirúrgico radical, ofreciendo así una alternativa terapéutica. De hecho, en casos aislados en los que se ha empleado imatinib en DFSP, se han obtenido elevadas tasas de respuesta, lo que sostiene la hipótesis de que las células del DFSP dependen de la expresión aberrante del PDGFRB para la proliferación y supervivencia celular 13. En cultivo de tejidos de DFSP se ha demostrado que el imatinib interfiere con el PDGFB1 2,14,15, y algunos autores sugieren que este fármaco induce a la apoptosis de las células tumorales pudiendo destruir totalmente el tumor 16, mientras otros piensan que produce una alteración del fenotipo del DFSP disminuyendo su índice de proliferación y en consecuencia su tamaño tumoral, pero no erradicándolo 17.
En conclusión, presentamos un caso de DFSP con componente de fibrosarcoma con una nueva translocación, no descrita previamente en la literatura médica (exón 19 CO1A1-exón 2 PDGFB). El gen de fusión COL1A1-PDGFB es específico del DFSP, por lo que su detección mediante técnicas de biología molecular puede tener un importante valor diagnóstico en los casos de diagnóstico diferencial difícil con otros sarcomas, y también puede constituir un indicador de tratamiento con inhibidores de la familia de las tirosincinasa, que pueden ser útiles en casos de DFSP localmente avanzados o metastásicos, o como tratamiento neoadyuvante previo a la cirugía, ofreciendo una nueva alternativa terapéutica farmacológica.
AGRADECIMIENTOS
A la Dra. Brufau por remitirnos el caso.
Declaración de conflicto de intereses Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Beatriz Llombart. Profesor Beltrán Báguena, 8.
46015 Valencia. España.
Beatriz.llombart@uv.es
Recibido el 28 de noviembre de 2005.
Aceptado el 24 de abril de 2006.
Estudio parcialmente realizado con la ayuda FIS P1040822.


