



La acropigmentación reticulada de Kitamura (APRK) es una genodermatosis infrecuente, incluida dentro de los trastornos pigmentarios reticulados congénitos, caracterizada por pigmentación acral lentiginosa, que ocurre principalmente en japoneses. El nevus de Ito es una melanocitosis dérmica, generalmente congénita, que afecta a la región acromioclavicular y deltoidea, más frecuente en mujeres asiáticas.
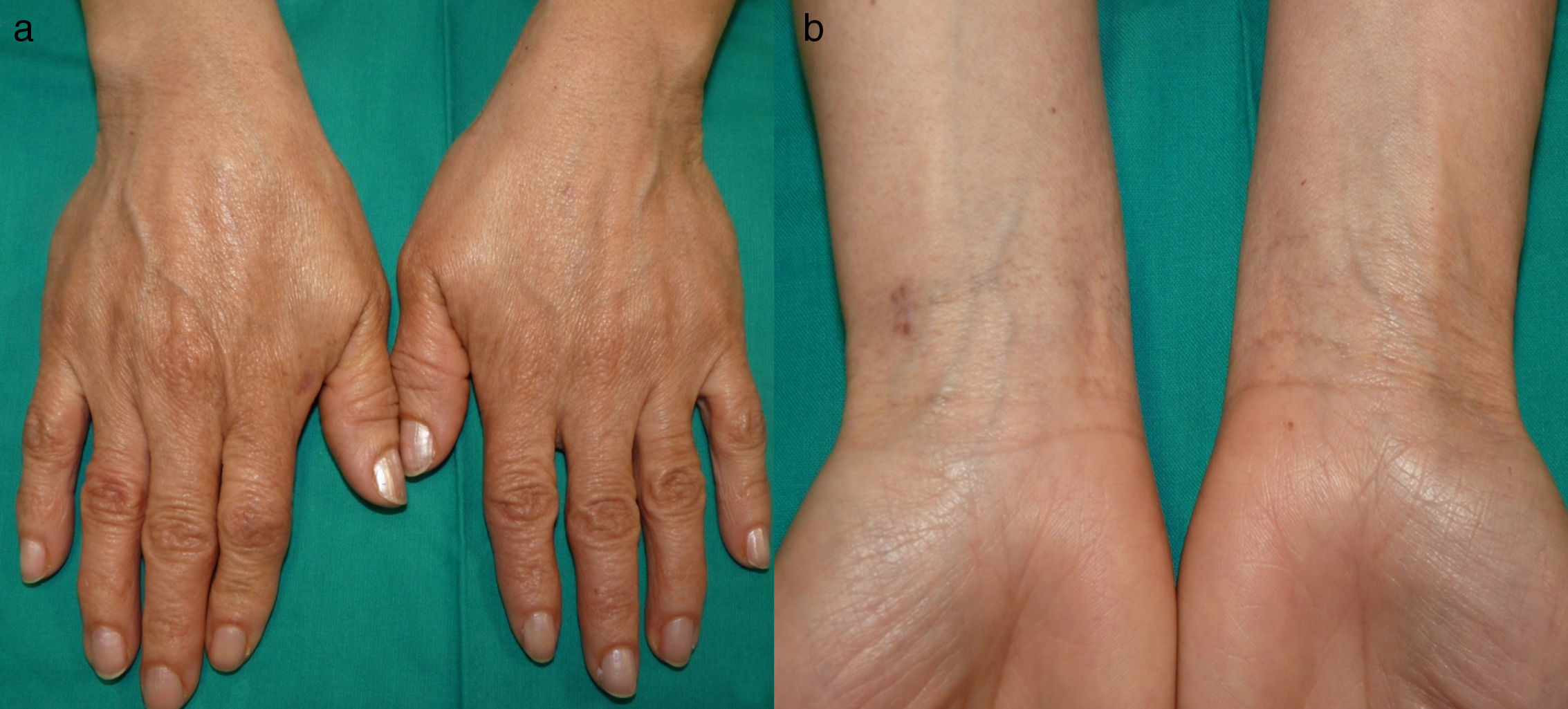
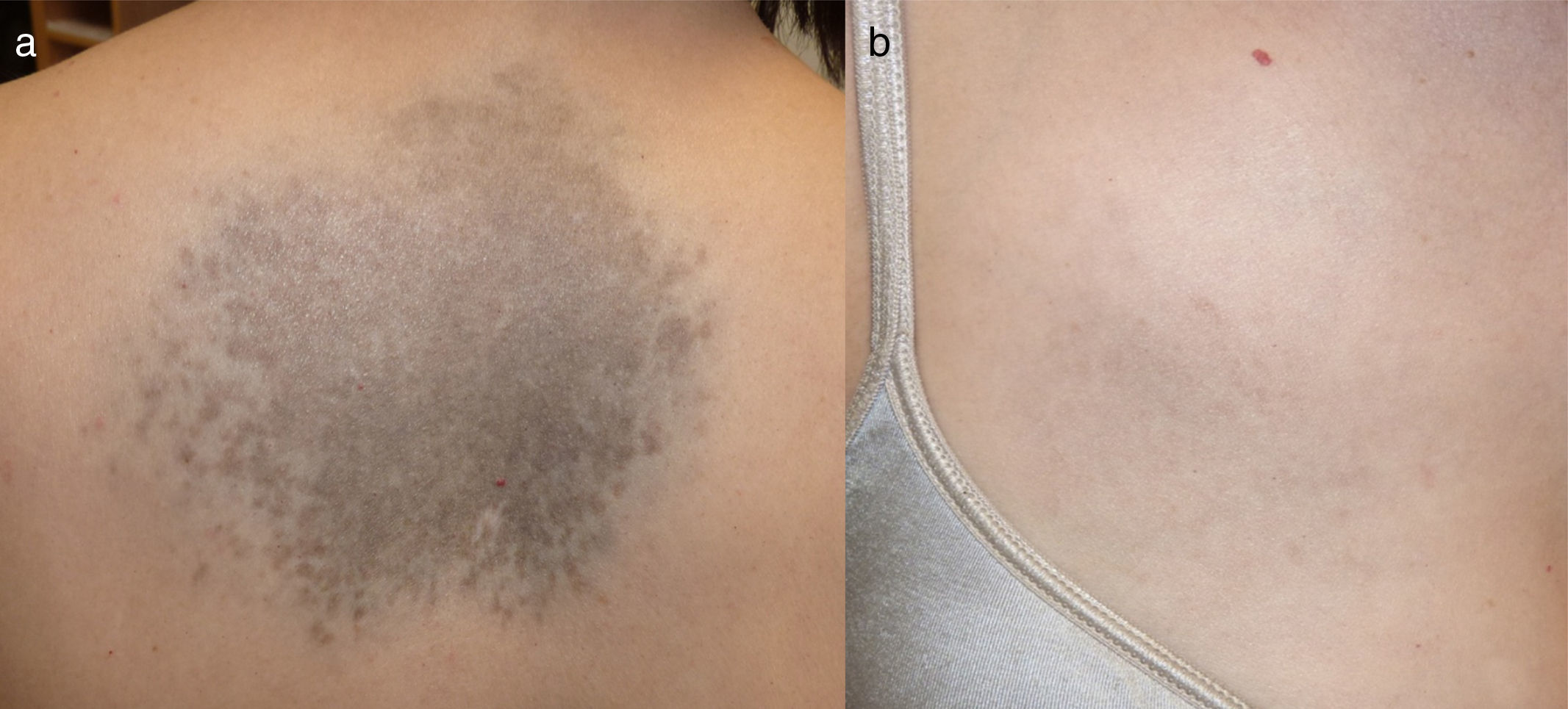
Una mujer de 47 años, sin antecedentes de interés, de fototipo iii, consultó por oscurecimiento lentamente progresivo y asintomático en el dorso de las manos y la cara desde la adolescencia. Refería que su madre tenía una pigmentación similar en las manos, pero no otros familiares (2 tías maternas, 2 hermanos, 3 hijos). Además, presentaba una lesión congénita y estable en la espalda y el hemitórax derecho. En la exploración se observaba una pigmentación lentiginosa bilateral y simétrica en el dorso de ambas manos, la cara anterior de las muñecas, los párpados y la región perilabial, acompañándose de pits palmoplantares e interrupción de dermatoglifos palmares (fig. 1). En la espalda, así como en el brazo y el hemitórax derecho, se observaba una placa azul-grisácea (fig. 2). La biopsia de la mano mostró una hiperplasia epidérmica lentiginosa con proyecciones bulbosas hiperpigmentadas de las crestas epidérmicas, y la del dorso, melanocitos dendríticos en la dermis (fig. 3). Con estos hallazgos el diagnóstico fue de APRK y nevus de Ito.
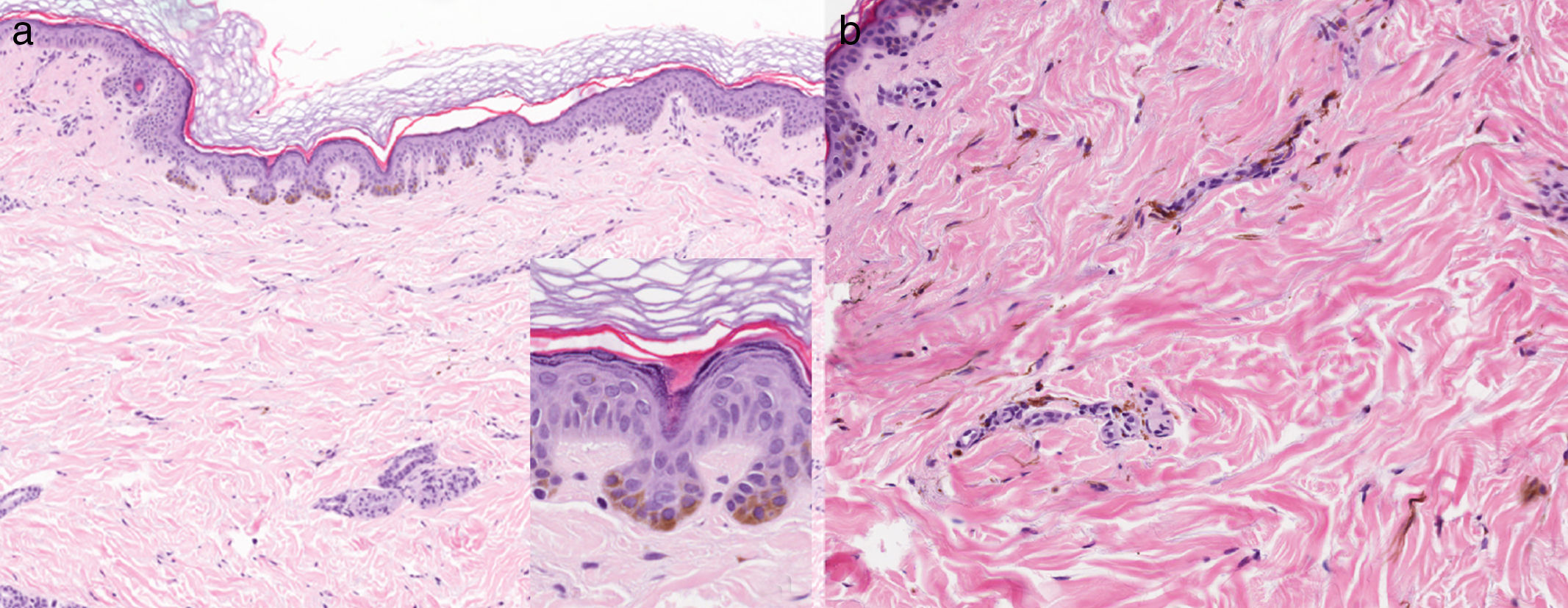
a. Biopsia de la muñeca que evidencia hiperplasia epidérmica lentiginosa con proyecciones bulbosas hiperpigmentadas de las crestas epidérmicas e incremento de los melanocitos basales (hematoxilina-eosina, ×10, y detalle en recuadro, ×40). b. Biopsia de la espalda con melanocitos dendríticos en la dermis papilar y reticular e hiperpigmentación de la capa basal epidérmica (hematoxilina-eosina, ×20).
Los trastornos pigmentarios reticulados comprenden un grupo de entidades congénitas y adquiridas con hiperpigmentación reticulada o tipo efélides y ocasional hipopigmentación, con confusión en la literatura en cuanto a la definición y los términos empleados. Las formas hereditarias son raras, de herencia variable, aunque generalmente autosómica dominante, con posibles anomalías asociadas, e incluyen, entre otros, disqueratosis congénita, enfermedad de Dowling-Degos, APRK, síndrome de Haber, acropigmentación reticulada de Dohi, síndrome de Naegeli-Franceschetti-Jadassohn, trastorno pigmentario reticular ligado al X y discromatosis universal hereditaria. En las formas adquiridas las lesiones suelen ser de mayor tamaño e incluyen la papilomatosis confluente y reticulada (Gougerot y Carteaud), el prurigo pigmentoso, el liquen plano pigmentoso, la melanosis de Riehl, el eritema ab igne, la cutis marmorata, la livedo reticularis y la pigmentación postinflamatoria, entre otros1.
La APRK es un trastorno poco frecuente, con aproximadamente 130 casos reportados2–7, más común en Japón, aunque su distribución es mundial, de herencia autosómica dominante, aunque existen casos esporádicos. Se manifiesta en la primera o segunda décadas, con máculas hiperpigmentadas reticuladas o tipo efélides, de 1-5mm, inicialmente atróficas, en el dorso de las manos y los pies, con extensión proximal y oscurecimiento progresivo agravado por la fotoexposición. Raramente afecta la cara y se acompaña de pits palmoplantares, interrupción de dermatoglifos y aisladamente se ha descrito alopecia localizada y ausencia de falanges terminales en los pies. Con dermatoscopia la pigmentación presenta una fina red de pigmento marrón reticular inespecífica y las depresiones palmoplantares, puntos marrones2. Histológicamente muestra atrofia epidérmica, elongación y aumento de melanina en las crestas interpapilares, con escaso infiltrado linfocítico perivascular y aumento del número de melanocitos basales positivos para DOPA4.
Existe controversia sobre si la APRK, la enfermedad de Dowling-Degos, el síndrome de Haber y la acropigmentación reticulada de Dohi son variantes de una misma enfermedad, ya que presentan solapamiento clínico-histológico (excepto la enfermedad de Galli-Galli, que muestra acantolisis suprabasal), principalmente la enfermedad de Dowling-Degos y la APRK3,4. La enfermedad de Dowling-Degos es autosómica dominante, afecta más a mujeres en la edad adulta, manifestándose con pigmentación reticulada y pápulas hiperqueratósicas marrones predominantemente en flexuras y tronco. Puede acompañarse de comedones en la cara y el cuello, pits periorales y cicatrices faciales, e incluso quistes epidérmicos, hidradenitis supurativa, queratosis seborreicas, quistes pilonidales y tumores como queratoacantomas y carcinomas espinocelulares. Histológicamente es similar a la APRK, pero con una mayor afectación folicular. Recientemente varios estudios genéticos apuntan que son trastornos distintos, pero no está claro. En la enfermedad de Dowling-Degos se han descrito mutaciones del gen de la citoqueratina 5 –KRT5– y, posteriormente, de los genes de las proteínas O-fucosiltransferasa 1 –POFUT1– y O-glucosiltransferasa 1 –POGLUT1–, presentando esta última mutación solapamiento clínico con la APRK8. En 2013 se identificaron varias mutaciones del gen ADAM10 en familias japonesas con APRK5 y, más recientemente, en otros casos6,7. ADAM10 codifica una desintegrina y metaloproteasa (ADAM), es un miembro de la familia ADAM (en inglés, a disintegrin and metalloprotease), la cual posee numerosas funciones biológicas y se expresa en la epidermis humana, células de melanoma y queratinocitos. Su mutación en ratones se asocia a pigmentación tipo pecas5.
Por otro lado, las melanocitosis dérmicas comprenden un grupo de trastornos congénitos benignos y menos frecuentemente adquiridos, caracterizados por la presencia de melanocitos dendríticos en la dermis. Son más comunes en mujeres asiáticas o africanas, siendo infrecuentes en la raza caucásica. Comprenden el nevus de Ota, de Ito, la mancha mongólica y el hamartoma melanocítico dérmico. La malignización de las melanocitosis dérmicas es muy rara, con apenas 4 casos en nevus de Ito9. Por otro lado, se ha descrito un caso de enfermedad de Dowling-Degos y melanoma amelanótico metastásico10. Hasta el momento, no se ha reportado APRK junto con otros trastornos melanocíticos. No se puede asegurar que la APRK y el nevus de Ito estén relacionados, ya que aunque ambos son trastornos pigmentarios, son distintos; así, en las melanocitosis dérmicas existe una proliferación de melanocitos dérmicos, mientras en la APRK se observa un aumento en el número de melanosomas en los queratinocitos y melanocitos lesionales con microscopia electrónica6. Hasta la fecha no se ha estudiado la mutación ADAM10 en las melanocitosis dérmicas.
Presentamos un caso de APRK y nevus de Ito en una mujer de raza caucásica, asociación no descrita hasta la fecha y ambas más frecuentes en asiáticos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos al Dr. Juan Luis Santiago Sánchez-Mateos su colaboración en este trabajo.


