


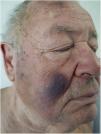

A 76-year-old man with a personal history of optic neuritis, cerebellar infarct, chronic moderate kidney failure, dyslipidemia, undergoing study for moderate pancytopenia with predominance of granulocyte and platelet series, which had appeared 2 years earlier (with a diagnosis of myelodysplastic syndrome) was referred to the dermatology department with bruise-like spots that had gradually appeared 6 months earlier on the face and back, with no associated pruritus.
Physical ExaminationThe physical examination revealed violaceous nodular plaques with poorly defined borders on the back, predominantly on the upper third, and on both cheeks (Fig. 1). Palpation revealed no locoregional enlarged lymph nodes in the neck, axillae, or groin, nor enlarged liver or spleen.
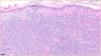
Histopathology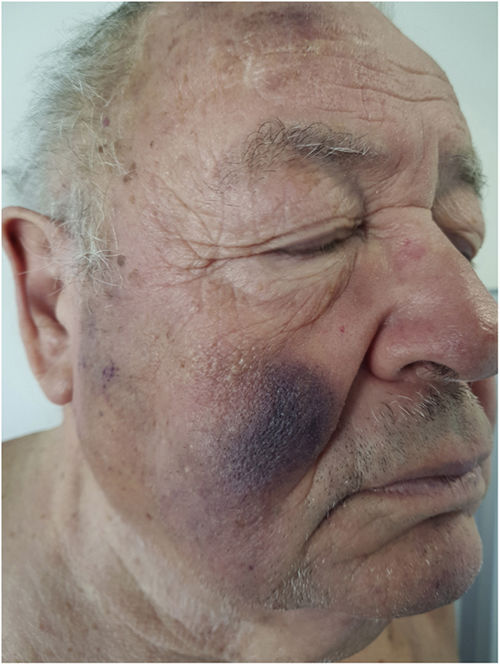
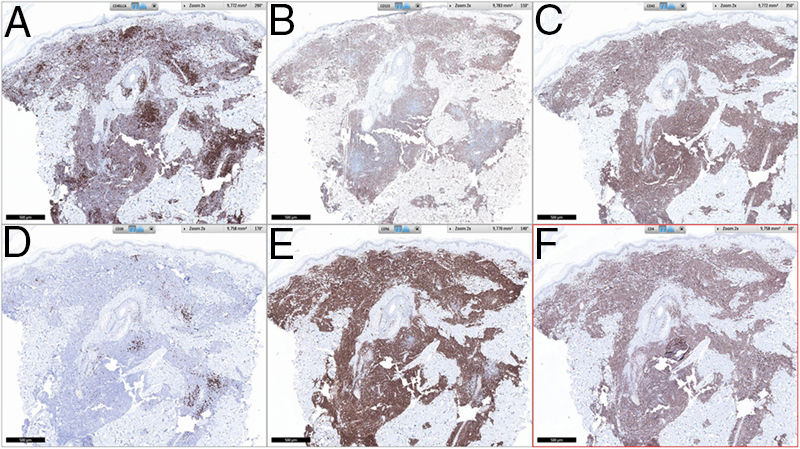
A lymphocytic infiltrate was observed throughout the thickness of the dermis, in the form of converging patches with a Grenz band against the epidermis (Fig. 2). Immune staining was positive for CD45, CD4, CD1a, CD43, CD56, and CD123 (Fig. 3), negative for CD20, CD138, CD8, and myeloperoxidase, and weakly positive for CD33. The Ki-67 labeling index was 40%.
Additional Tests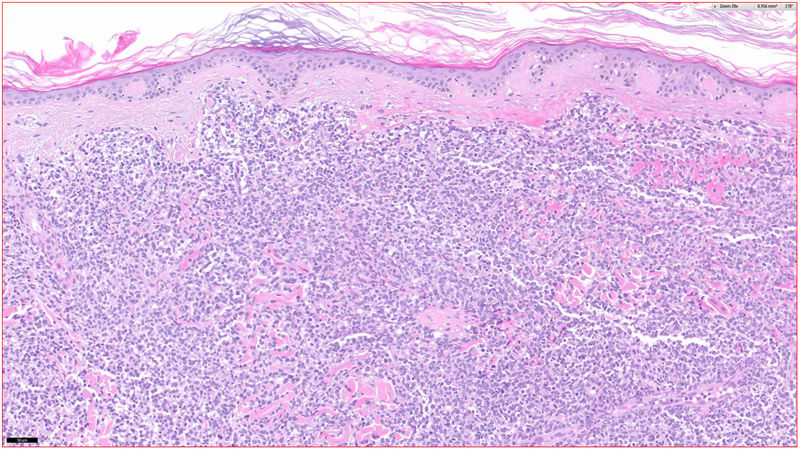
Analyses revealed the following: Hb, 11.9g/dL; l=2180/Ul; platelets, 78.000/Ul; and B2 microglobulin, 6.7 (N<5mg/dL). The immune phenotype of the peripheral blood cells was as follows: monocytes, 16.5%; lymphocytes, 28.16%; TCD3+ lymphocytes, 22.5%; CD4+, 15.8%; CD8+, 5.66%; B lymphocytes, 0.6%; NK cells, 4.4%; and neutrophils, 53.88%. All cells were mature CD10+ granulocytes. No blast cells were detected. Fine-needle aspiration of the bone marrow revealed normal bone-marrow cells with erythroid hyperplasia with no evident dysplastic signs of blood disease. Imaging studies produced normal results (abdominal ultrasound, CT scan, and PET-CT).
What is your diagnosis?Diagnosis
Blastic plasmacytoid dendritic cell neoplasm (WHO 2017) or CD4+/CD56+ hematodermal neoplasm.
Clinical Course and TreatmentStudy of disease extension revealed no involvement of the bone marrow. Treatment was instated by protocol with cyclophosphamide, vincristine, doxorubicin, and dexamethasone for frail patients over 45 years of age. The clinical course was favorable after 3 months of treatment and no new lesions appeared. The patient continues in periodic follow-up by the dermatology and hematology departments.
CommentDifferent subtypes of lymphoma that express CD56 have emerged in recent years. These include hematodermal CD4+/CD56+ or blastic plasmacytoid dendritic cell neoplasm. This entity was first described by Adachi in 1994.1 In 2001, Chaperot2 considered plamacytoid dendritic cells to be the precursor cells of this neoplasm and the entity acquired its definitive name in the 2005 WHO-EORTC update.3 It consists of an aggressive hematologic disorder with a high incidence of cutaneous involvement and a major risk of leukemic cell dissemination.4
It typically affects middle-aged and elderly men with no predominance of any race. It rarely presents in children and only 12 pediatric cases have been reported to date.5 It presents clinically as violaceous bruise-like spots or plaques, which are painless in most cases, with a predilection for the torso, although the head and neck may also be involved, as may the limbs. Involvement of the lymph nodes is relatively common (between 40% and 50% of cases), although B symptoms are extraordinarily rare.6
From a histology point of view, this variety of lymphoma does not present epidermotropism. The lymphocytic infiltrate affects the dermis massively and sometimes goes deeper into the subcutaneous cellular tissue. Necrosis and vascular invasion are not usual and mitotic figures tend to be present
The immune-staining profile of most of these tumors is the following: CD4+/CD56+/CD8–/CD7–/CD45+ and negative for B-cell markers and other T cells.5
The differential diagnosis should focus on lymphoproliferative disorders with primary and secondary skin involvement, especially acute leukemia, myelodysplastic syndromes, and lymphoblastic lymphomas and nasal or extranasal NK lymphomas.6
Prognosis is poor and the mean survival rate has been estimated in most series consulted to be approximately 14 months. Dissemination to the bone marrow tends to be a rapid process once the skin lesions appear. For this reason, most of these cases must be treated at multidisciplinary centers and with aggressive regimens that consider this entity as equivalent to an acute leukemia. In cases where response is favorable, development of residual hyperpigmentation is typical, as is recurrence if therapy is interrupted. A good response has been reported following use of the combined protocol of chemotherapy and radiation therapy followed by allogenic stem-cell transplant.7
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Ruiz-Villaverde R, Rueda-Villafranca B, Ruiz-Carrascosa JC. Manchas contusiformes de aparición progresiva en la cara y la espalda. Actas Dermosifiliogr. 2021;112:447–448.


