


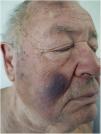

Un hombre de 76 años, con antecedentes personales de neuritis óptica, infarto cerebeloso, insuficiencia renal crónica moderada, dislipidemia, en estudio por una pancitopenia moderada con predominio de las series granulocíticas y plaquetaria de 2 años de evolución (con diagnóstico de síndrome mielodisplásico) fue remitido a la consulta de dermatología por presentar un cuadro cutáneo de manchas contusiformes, de aparición progresiva localizadas en la cara y la espalda, sin prurito asociado, de 6 meses de evolución.
Exploración físicaEl examen clínico reveló unas placas nodulares violáceas de bordes mal delimitados en la espalda, con predominio en el tercio superior y en ambas mejillas (fig. 1). No se palparon adenopatías locorregionales en las cadenas cervicales, axilares ni inguinales, ni hepatoesplenomegalia.
Histopatología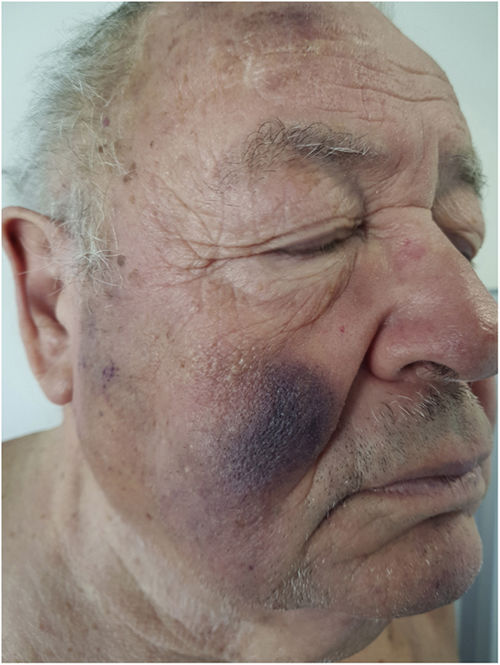
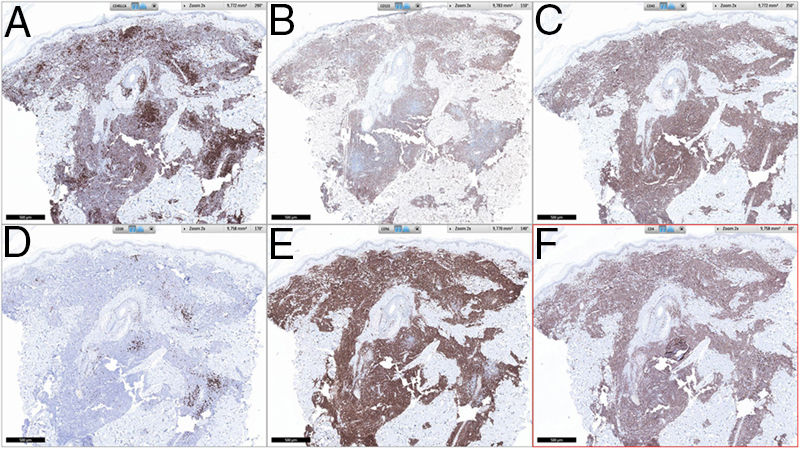
Se observó un infiltrado linfocitario en todo el espesor de la dermis, a modo de parches confluentes con una banda Grenz de respeto epidérmico (fig. 2). Mediante técnicas inmunohistoquímicas se demostró una positividad para CD45, CD4, CD1a, CD43, CD56 y CD123 (fig. 3), resultando negativas para CD20, CD138, CD8 y mieloperoxidasa y débilmente positivas para CD33. El índice Ki67 fue de un 40%.
Pruebas complementarias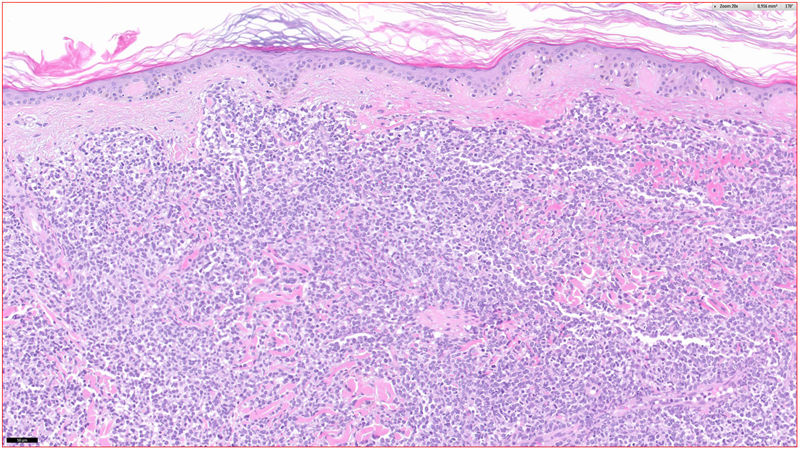
Los estudios analíticos revelaron los siguientes hallazgos: Hb 11,9g/dl, l=2180/Ul, plaquetas 78.000/Ul y B2 microglobulina 6,7 (N<5mg/dl). El inmunofenotipo de las células de sangre periférica mostró: monocitos 16,5%, linfocitos 28,16%, linfocitos TCD3+: 22,5%, CD4+: 15,8%, CD8+: 5,66%, linfocitos B: 0,6%, células NK 4,4% y neutrófilos 53,88%. Todas las células eran granulocitos maduros CD10+. No había detección de células blásticas. La punción-aspiración de la médula ósea fue informada de médula normocelular, con una hiperplasia eritroide sin rasgos displásicos evidentes de hemopatía. Las pruebas de imagen ofrecieron resultados dentro de la normalidad (ecografía abdominal, TAC y PET-TAC).
¿Cuál es su diagnóstico?DiagnósticoSe estableció el diagnóstico de neoplasia blástica de células dendríticas plasmocitoides (OMS 2017)-CD4+/CD56+ neoplasia hematodérmica.
Evolución y tratamientoEl estudio de extensión no evidenció una afectación de la médula ósea. Se inició tratamiento mediante protocolo con ciclofosfamida, vincristina, doxorrubicina y dexametasona para pacientes mayores de 45 años frágiles. La evolución fue buena a los 3 meses del inicio del mismo, y sin la aparición de nuevas lesiones cutáneas. Continúa en revisión periódica por los servicios de dermatología y hematología.
ComentarioEn los últimos años han emergido diferentes subtipos de linfomas que expresan CD56. Dentro de ellos se ha descrito la neoplasia hematodérmica CD4+/CD56+ o neoplasia blástica de células dendríticas plasmocitoides. Esta entidad fue inicialmente descrita por Adachi en 19941. En 2001 Chaperot2 consideró las células dendríticas plasmocitoides como las células precursoras de esta neoplasia, y en la revisión de la WHO-EORTC publicada en 2005 adquirió su denominación definitiva3. Consiste en un desorden hematológico agresivo con una incidencia alta de afectación cutánea y un importante riesgo de diseminación leucémica4.
Típicamente afecta a hombres de edad media o avanzada sin un predominio por la raza. Es rara su presentación pediátrica y solo se han descrito 12 casos hasta el momento5. Se presenta clínicamente mediante manchas o placas de aspecto contusiforme, violáceas y, en la mayoría de los casos, indolentes, con una predilección por el tronco, si bien puede existir compromiso de la cabeza y el cuello, así como de las extremidades. La afectación de los ganglios linfáticos es relativamente común (40-50% de los casos), si bien la presencia de síntomas B es extraordinariamente infrecuente6.
Desde el punto de vista histológico esta variedad de linfoma no presenta epidermotropismo. El infiltrado linfocitario afecta de forma masiva a la dermis y, en ocasiones, profundiza en el tejido celular subcutáneo. No suele existir necrosis ni invasión vascular y las figuras mitóticas suelen estar presentes.
El perfil inmunohistoquímico de la mayoría de estos tumores es: CD4+/CD56+/CD8–/CD7–/CD45+ y negativo para los marcadores de células B y otras células T5.
El diagnóstico diferencial debe centrarse en aquellos trastornos linfoproliferativos con una afectación cutánea primaria y secundaria, en especial con la leucemia aguda, los síndromes mielodisplásicos y los linfomas linfoblásticos, los linfomas NK nasales o extranasales6.
El pronóstico es malo y la media de supervivencia se ha estimado en la mayoría de las series consultadas en torno a los 14 meses. La diseminación a la médula ósea suele ser un proceso rápido una vez que han aparecido las lesiones cutáneas. Es por ello que la mayoría de estos casos deben ser tratados en centros multidisciplinares y con regímenes terapéuticos agresivos que consideren esta entidad equivalente a una leucemia aguda. En aquellos casos donde hay respuesta favorable es típico el desarrollo de hiperpigmentaciones residuales y la recidiva si se interrumpe la terapia. Se ha comunicado una respuesta buena tras el uso de protocolo combinado de quimio y radioterapia seguido de trasplante alogénico de células madre7.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


