



El xantogranuloma juvenil es una histiocitosis de células no Langerhans que acontece en la edad infantil; sin embargo, se han descrito varios casos en el adulto, algunos de ellos en relación con hemopatías malignas. Presentamos el caso de una mujer de 61 años de edad con lesiones diseminadas de xantogranuloma juvenil que a los 4 años de seguimiento desarrolló un linfoma de tipo folicular. Tras 6 meses de tratamiento con quimioterapia y rituximab se consiguió remisión del linfoma y la involución de las lesiones cutáneas. Destacamos este caso por tratarse de una entidad poco frecuente y de difícil diagnóstico en el adulto, así como por ser el primer caso asociado con linfoma folicular y que ha presentado una excelente respuesta con quimioterapia y rituximab. Además, dada su posible asociación con enfermedades hematológicas, el xantogranuloma juvenil podría representar una manifestación de una neoplasia oculta.
Juvenile xanthogranuloma is a non-Langerhans cell histiocytosis that typically affects children, but several cases have been reported in adults, some in connection with hematologic malignancies. We present the case of a 61-year-old woman with multiple xanthogranulomas who developed a follicular lymphoma after 4 years of follow-up. After 6 months of treatment with chemotherapy and rituximab, the cutaneous lesions disappeared and the patient achieved remission from lymphoma. We highlight this case because xanthogranuloma is a rare disorder that is difficult to diagnose in adults and also because this is the first report of an association between xanthogranuloma and follicular lymphoma. Excellent response was achieved with chemotherapy and rituximab. Finally, given the possible association between xanthogranulomas and hematologic diseases, these lesions may be a cutaneous manifestation of an occult malignancy.
El xantogranuloma juvenil (XGJ) es una histiocitosis de células no Largerhans (HCNL) que afecta a niños en los primeros años de la vida y se caracteriza por múltiples lesiones papulonodulares, pardo-amarillentas y autorresolutivas. Histológicamente son característicos los histiocitos y las células multinucleadas tipo Touton. Los casos publicados en el adulto son escasos y consisten, mayoritariamente, en lesiones solitarias que no suelen regresar espontáneamente1. Su conocimiento ha despertado interés, dada la posibilidad de asociarse con enfermedades oncohematológicas.
Caso clínicoMujer de 61 años con antecedentes de infección tuberculosa latente hacía 30 años, fumadora ocasional. Se le había practicado un legrado uterino secundario a miomas que requirió de hemotransfusión. En el año 2004 comenzó con lesiones papulosas de color pardo-amarillento en el tronco, cuyo diámetro oscilaba entre 3 y 5mm, que fueron aumentando en número y extendiéndose progresivamente por el abdomen y la raíz de los miembros (fig. 1A y B).

El estudio histológico de una de las lesiones evidenció un infiltrado que se distribuía predominantemente por la dermis media, de forma difusa, disecando haces de colágeno y formando agregados en la dermis profunda. Dicho infiltrado se constituía fundamentalmente por células de citoplasma amplio y hábito histiocitario, junto con células gigantes multinucleadas, algunas de ellas tipo Touton; además se observaban algunos linfocitos sin atipias y eosinófilos entremezclados (figs. 2 y 3). La inmunohistoquímica fue positiva para células de estirpe monocítica-macrofágica (CD68) y negativa para células de Langerhans (proteína S-100 y CD1a). Con el diagnóstico de XGJ del adulto fue seguida en consultas con aparición progresiva de nuevas lesiones clínicamente similares. A los 4 años se objetivó una adenopatía inguinal izquierda de crecimiento rápido, sin sintomatología acompañante. La extirpación y el estudio histológico de la misma mostró un proceso linfoproliferativo nodular constituido por una celularidad centrocítica con fenotipo centrogerminal (la inmunohistoquímica reveló positividad frente a CD20, CD23 y CD10 en áreas foliculares, con expresión difusa de bcl2 y selectiva de bcl6 en folículos), la cual mostró reordenamiento clonal del gen IGH (fig. 3). En el estudio de extensión destacaron adenopatías retroperitoneales y perivasculares, con una adecuada diferenciación de las 3 series hematopoyéticas, sin infiltración neoplásica en la biopsia de médula ósea. Todos los hallazgos anteriores condujeron al diagnóstico final de linfoma B folicular bien diferenciado y de bajo grado, en estadio iiia.

.")
A. H-E×100. Infiltrado de células de citoplasma amplio y hábito histiocitario, algunas células gigantes multinucleadas, junto con linfocitos sin atipias y escasos eosinófilos. B. Inmunohistoquímica para CD1a×200. Obsérvese la negatividad de las células. C. Inmunohistoquímica para CD68×200. Se observa positividad granular en los citoplasmas celulares. D. H-E×100. Estudio histológico de ganglio linfático inguinal. Se observa una proliferación linfoide centrocítica (células pequeñas hendidas).
Un nuevo estudio histológico de otra de las lesiones cutáneas, que habían aumentado en número, mostró hallazgos superponibles a la primera biopsia, sin reordenamiento del gen IGH debido a que no había material genético suficiente. Se instauró tratamiento con fludarabina 40mg, ciclofosfamida 250mg 3 días y rituximab 375mg/m2 en 4 ciclos mensuales, completándose posteriormente con 4 ciclos de rituximab 375mg/m2 semanal. A los 6 meses de finalizar el tratamiento se observó una remisión completa de la neoplasia, así como una involución de las lesiones cutáneas que dejaron cicatrices deprimidas e hiperpigmentadas (fig. 1C). En el momento actual, tras 2 años de seguimiento, no hay indicios de recidiva hematológica ni nueva aparición de lesiones cutáneas.
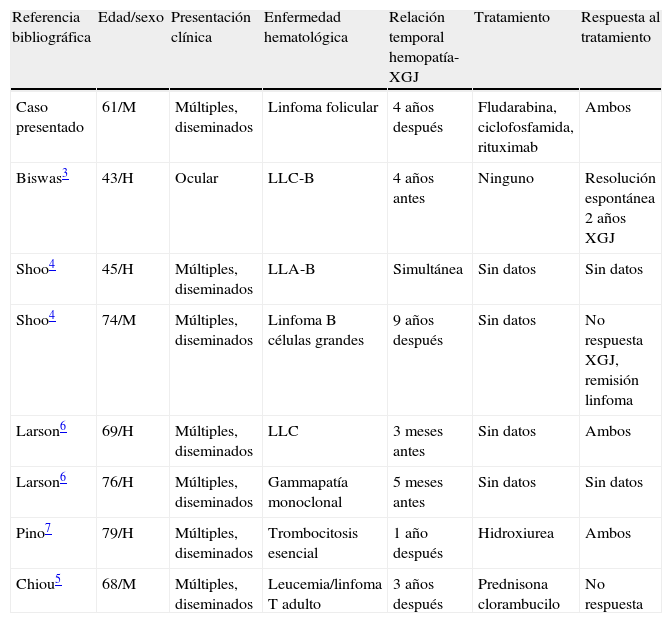
DiscusiónEl XGJ del adulto se describió por primera vez en 1963, habiéndose comunicado hasta el momento unos 125 casos1,2, siendo en su mayoría lesiones solitarias. Sin embargo, se han descrito 21 casos con lesiones diseminadas con número mayor de 101–6, presentando hallazgos histológicos semejantes a los del niño. De todos los casos de XGJ del adulto revisados, 7 se relacionaron con una enfermedad hematológica maligna3–7 (tabla 1), representando las leucemias3–5 el grupo más importante con 3 casos, seguido por 2 pacientes con linfomas3,5, un caso de gammapatía monoclonal6 y otro de trombocitosis esencial7. No hemos encontrado casos de XGJ que se relacionen con linfoma folicular, por lo que nuestro paciente sería el primero descrito. En todos los pacientes en los que el XGJ se relacionó con una discrasia sanguínea los pacientes presentaron lesiones múltiples con afectación exclusivamente cutánea, excepto un caso con afectación periocular3. El XGJ se presentó previa, simultánea o posteriormente al diagnóstico de la hemopatía, con un periodo que osciló entre los 5 meses y los 9 años.
Casos de xantogranuloma juvenil en el adulto asociados a hemopatías
| Referencia bibliográfica | Edad/sexo | Presentación clínica | Enfermedad hematológica | Relación temporal hemopatía-XGJ | Tratamiento | Respuesta al tratamiento |
| Caso presentado | 61/M | Múltiples, diseminados | Linfoma folicular | 4 años después | Fludarabina, ciclofosfamida, rituximab | Ambos |
| Biswas3 | 43/H | Ocular | LLC-B | 4 años antes | Ninguno | Resolución espontánea 2 años XGJ |
| Shoo4 | 45/H | Múltiples, diseminados | LLA-B | Simultánea | Sin datos | Sin datos |
| Shoo4 | 74/M | Múltiples, diseminados | Linfoma B células grandes | 9 años después | Sin datos | No respuesta XGJ, remisión linfoma |
| Larson6 | 69/H | Múltiples, diseminados | LLC | 3 meses antes | Sin datos | Ambos |
| Larson6 | 76/H | Múltiples, diseminados | Gammapatía monoclonal | 5 meses antes | Sin datos | Sin datos |
| Pino7 | 79/H | Múltiples, diseminados | Trombocitosis esencial | 1 año después | Hidroxiurea | Ambos |
| Chiou5 | 68/M | Múltiples, diseminados | Leucemia/linfoma T adulto | 3 años después | Prednisona clorambucilo | No respuesta |
Adaptada de Shoo et al.4. H: hombre; LLA-B: leucemia linfática aguda de células B; LLC: leucemia linfática crónica; M: mujer; XGJ: xantogranuloma juvenil.
La respuesta del XGJ y del proceso tumoral tras el tratamiento dirigido de la neoplasia sanguínea fue variable, lográndose remisión del proceso tumoral e involución de las lesiones cutáneas en solo dos casos6,7.
En la edad pediátrica algunos estudios han visto incrementado el riesgo de desarrollar leucemias cuando hay XGJ y neurofibromatosis, lo cual ha sido cuestionado posteriormente3. Se han publicado 2 casos pediátricos de XGJ con afectación cutánea diseminada relacionados con enfermedades mieloproliferativas. Un varón de 3 años 6 meses después de presentar un XGJ desarrolló una LLA-B, sin alcanzar remisión de la leucemia tras la quimioterapia, aunque sí un ligero aplanamiento de las lesiones cutáneas8. Un neonato con XGJ y síndrome hemagofagocítico desarrolló una leucemia mielomonocítica crónica juvenil9. Por otro lado, Castro et al.10 recogieron 3 niños con edades comprendidas entre los 8 y los 13 años con lesiones de XGJ de localización inusual, afectando ganglios linfáticos, huesos o pulmón, los cuales aparecieron junto con una LLA, con intervalos entre la aparición del XJ y la LLA que oscilaron entre uno y 12 años. No obstante, dada la mayor incidencia de esta enfermedad en los niños, su curso autorresolutivo y la descripción de formas de presentación atípica, nos lleva a pensar que el XGJ presenta una menor relación con neoplasias sanguíneas en la edad pediátrica.
Desde el punto de vista etiopatogénico se especula que las histiocitosis se desarrollarían a partir de la proliferación de un precursor común CD34+ de la médula ósea. La multiplicación anómala de estas células se debería a la acción de distintos estímulos, entre los que podrían encontrarse citoquinas o gammaglobulinas producidas por un proceso tumoral subyacente11. Por otra parte, se ha propuesto que algunas histiocitosis de células de Langerhans relacionadas con enfermedades mielo o linfoproliferativas podrían tener un origen común en una célula hematopoyética precursora con capacidad para desdiferenciarse tanto en células tumorales, que darían lugar a la neoplasia, como en células de la línea monocítica-dendrítica, que originaría la histiocitosis12. En este sentido se ha descrito recientemente un XGJ de afectación visceral (hígado y bazo) en un niño de 5 años, 3 meses después del diagnóstico de una LLA-T, que mostró un reordenamiento clonal bi-alélico entre el TCRγ de los blastos y el de los histiocitos13. No se logró establecer ningún tipo de relación clonal entre las células linfoproliferativas y los histiocitos que formaban parte de las lesiones cutáneas de nuestro paciente, debido a la falta de material genético de la biopsia cutánea. Así mismo, no hemos encontrado ningún otro caso de XGJ donde se muestre reordenamiento clonal entre la población tumoral hematológica ni la histiocitosis, por lo que sería interesante poder demostrar esta relación en futuros pacientes que presentasen ambas patologías; estudios que serían extensibles a otros casos de discrasias sanguíneas que se han relacionado con distintos tipos de HCNL, como el xantoma diseminado o el xantogranuloma necrobiótico.
En relación con el tratamiento no existe ningún régimen terapéutico que haya demostrado ser curativo para el XGJ diseminado del adulto. Los pacientes que presentaron enfermedad tumoral y respuesta de las lesiones cutáneas lo hicieron a la terapia antineoplásica. Nuestro paciente recibió tratamiento quimioterápico con fludarabina y ciclofosfamida unido a rituximab (anticuerpo monoclonal anti-CD20 que inhibe los linfocitos CD20+). El tratamiento citostático provocaría la apoptosis de las células tumorales, eliminando consecuentemente la producción de las distintas citoquinas y otros mediadores que podrían haber intervenido en la proliferación de los histiocitos. Se han propuesto distintos mecanismos por los cuales el rituximab inhibiría a los linfocitos, entre los que destaca la modulación que realizaría a través del sistema macrofágico-histicoitario14, lo cual podría haber contribuido a la resolución del XGJ en nuestro caso. No hemos encontrado ningún caso de XGJ diseminado tratado con rituximab, aunque sí se ha comunicado respuesta completa en un xantogranuloma necrobiótico asociado con un linfoma linfocítico con el mismo esquema terapéutico que nuestra paciente15.
ConclusiónSe presenta el primer caso de XGJ diseminado del adulto asociado con linfoma folicular con excelente respuesta al tratamiento citostático y rituximab, lo que apoyaría su posible asociación con neoplasias hematológicas. Aunque son pocos los casos comunicados para considerar esta entidad como un verdadero síndrome paraneoplásico, es aconsejable un seguimiento y despistaje de tumores, sobre todo hematológicos, siendo necesaria una exploración general y estudios complementarios dirigidos a aquellos pacientes con lesiones numerosas y de aparición progresiva.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


