


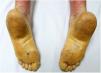
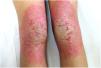
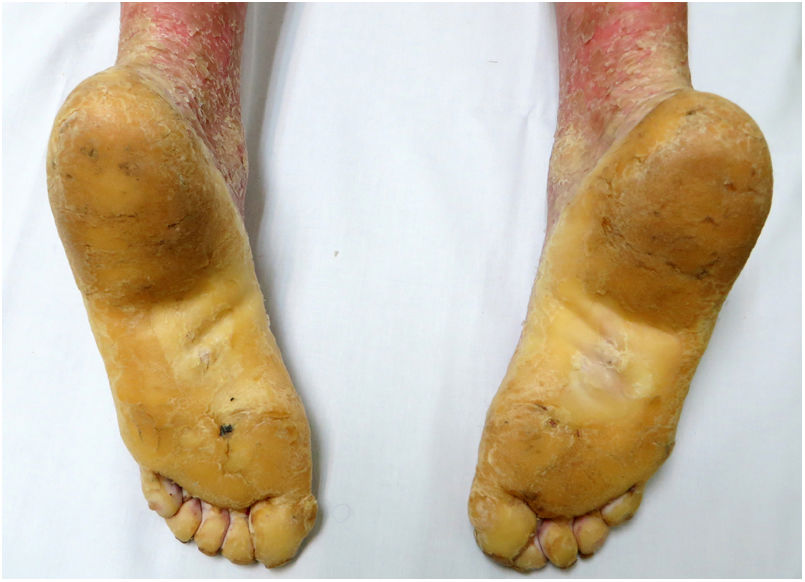
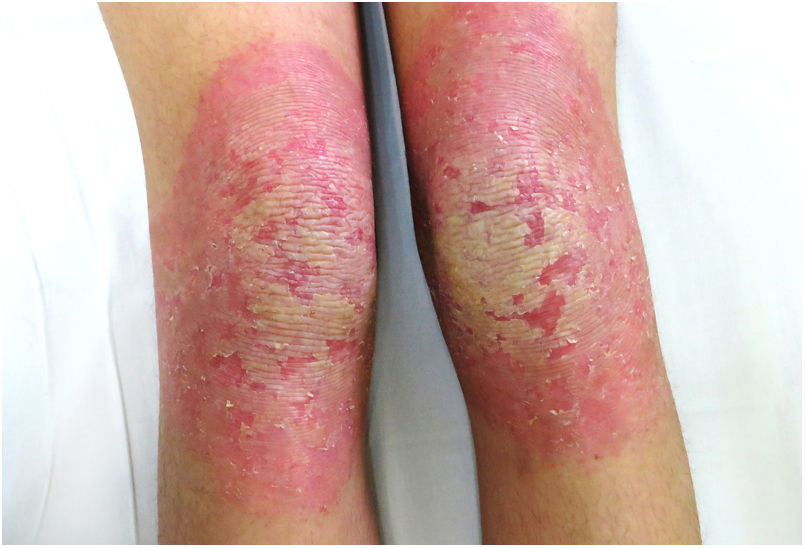
An 18-year-old woman from Morocco presented with diffuse waxy keratoderma on the palms and soles that she had suffered since the age of 3 years (Fig. 1), accompanied by well-defined erythematous scaly plaques on the knees and the dorsal aspect of the hands and feet (Fig. 2). She also presented flexion contractures and pseudoainhum on the fingers.
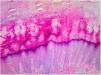
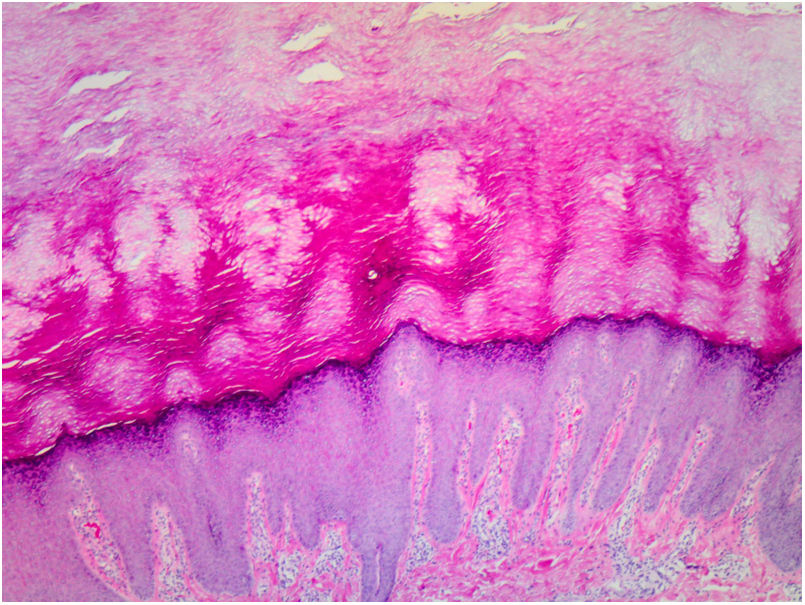
Histopathology of the lesions revealed marked orthokeratotic hyperkeratosis, acanthosis, and increased thickness of the stratum granulosum (Fig. 3).
The patient, whose parents were consanguineous, had a twin sister with the same condition and 3 other healthy brothers. A genetic study detected the c.82delT mutation in the SLURP-1 gene on chromosome 8q24.3.
What Is Your Diagnosis?
DiagnosisMal de Meleda.
CommentsThe lesions were clinically and histologically compatible with Mal de Meleda, and the diagnosis was confirmed by a genetic study. Treatment with 9% salicylic petrolatum, topical zinc sulfate, and oral acitretin (20mg/d) were prescribed. However, the patient did not return to the clinic and therefore her evolution remains unknown.
Mal de Meleda is a rare autosomal recessive genodermatosis caused by a mutation in the SLURP-1 (secreted lymphocyte antigen 6/urokinase receptor-related protein 1) gene on chromosome 8q24.3. SLURP-1 protein is expressed in the stratum granulosum, where it regulates epidermal homeostasis and inhibits TNF-α secreted by macrophages. The mutation in this gene results in epidermal hyperproliferation and inflammation.1,2
The disease is characterized by the appearance during the first 3 years of life of a diffuse palmoplantar hyperkeratosis that extends to the dorsal aspect of the hands and feet (transgrediens)4,5 and to the knees, elbows, and perioral region, and it tends to worsen with time (progrediens). It is accompanied by hyperhidrosis and bromhidrosis due to maceration and frequent superinfections. It is also associated with nail alterations, sclerodactyly, digital constrictions (pseudoainhum), and joint contractures, and exerts a marked impact on the quality of life of affected patients.1–6
It was first described on the island of Mljet (Meleda), Croatia, where reproductive isolation and consanguinity favored the transmission and increased incidence of this recessive disease. Cases were subsequently described in North Africa and the Middle East, following the commercial routes of the Medieval Republic of Dubrovnik.3 Currently, the estimated prevalence in the general population is 1 per 100000 inhabitants.2
In the present case, the genetic study revealed that the patient was a homozygous carrier of the pathogenic c.82delT variant in the SLURP-1 gene, which is associated with this disease.1–5 However, based on the available data we cannot rule out the possibility that the patient is a double heterozygous carrier of this mutation and a deletion in SLURP-1. To confirm homozygosis, analyses of the mutation of interest in the parents would be required to determine whether both of them are heterozygous carriers.
Therapeutic options include emollients, topical keratolytics, antibiotic therapy to treat superinfections, grafts, and systemic retinoid treatment, which is the only treatment with proven efficacy, improving epidermal thickening and contractures but not erythema.2,4
Mal de Meleda is a rare form of palmoplantar keratoderma, but has a highly characteristic clinical presentation, which should be borne in mind.
Conflicts of InterestThe authors declare that they have no conflicts of interest.


