



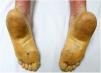
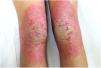
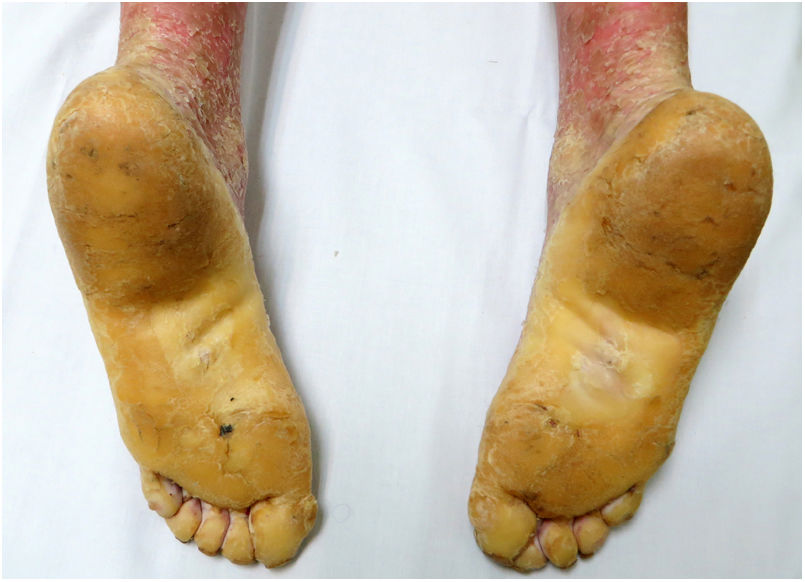
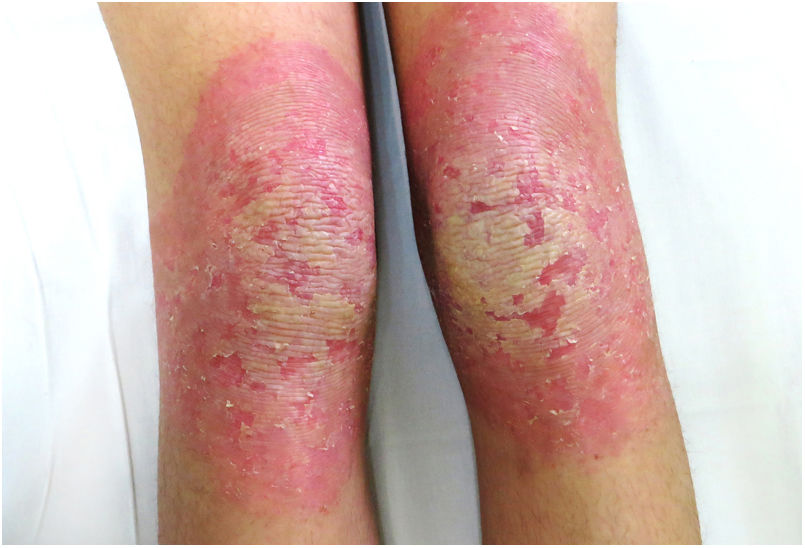
Una mujer de 18 años, natural de Marruecos, consultó porque desde los tres años de edad presentaba una queratodermia cérea difusa en las palmas y las plantas (fig. 1), acompañada de unas placas eritematodescamativas, bien delimitadas, en el dorso de las manos y los pies, así como en las rodillas (fig. 2). Presentaba, además, contracturas de flexión en los dedos de las manos y pseudoainhum.
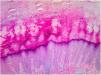
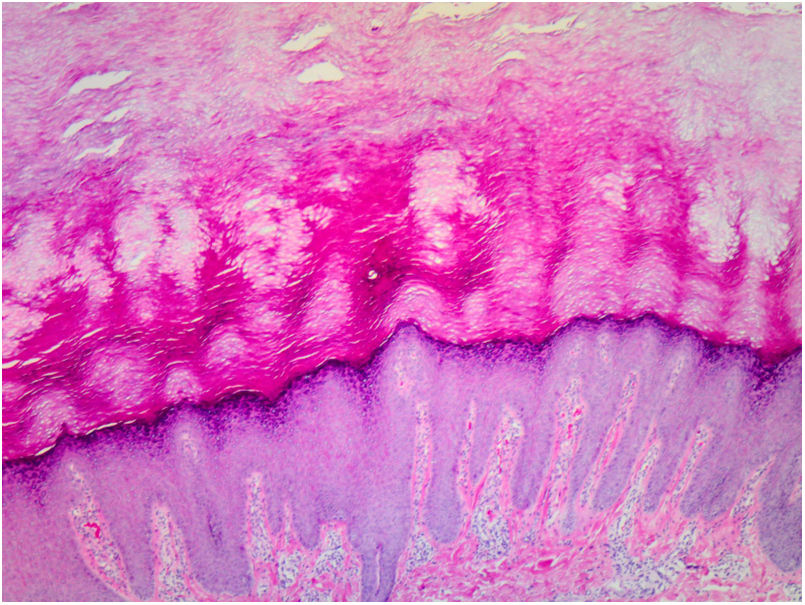
La anatomía patológica de las lesiones mostró una marcada hiperqueratosis ortoqueratósica, acantosis y un aumento del espesor del estrato granular (fig. 3).
Tenía una hermana gemela con la misma condición y otros tres hermanos sanos, existiendo relación de consanguinidad entre sus progenitores. El estudio genético detectó la mutación c.82delT en el gen SLURP-1 en el cromosoma 8q24.3.
¿Cuál es el diagnóstico?
DiagnósticoMal de Meleda.
ComentariosLas lesiones resultaron clínica e histopatológicamente compatibles con el mal de Meleda, y el estudio genético confirmó el diagnóstico. Se pautó tratamiento con vaselina salicílica al 9%, sulfato de zinc tópico y acitretina oral a dosis de 20mg/día, pero se desconoce la evolución, dado que la paciente no volvió a consulta.
El mal de Melda es una genodermatosis rara autosómica recesiva causada por la mutación del gen SLURP-1 (proteína 1 relacionada con el receptor activador de plasminógeno de tipo urocinasa/antígeno 6 secretado por los linfocitos) en el cromosoma 8q24.3. La proteína SLURP-1 se expresa en el estrato granuloso, regulando la homeostasis epidérmica e inhibiendo el α-TNF secretado por los macrófagos, por lo que la mutación de su gen produce una hiperproliferación epidérmica e inflamación1,2.
Cursa con la aparición durante los tres primeros años de vida de una hiperqueratosis palmoplantar difusa con extensión al dorso de las manos y los pies (transgrediens)4,5 y a las rodillas, los codos y la región perioral (progrediens). Se acompaña de hiperhidrosis y bromhidrosis, debido a la maceración y a las sobreinfecciones frecuentes. Además, asocia alteraciones ungueales, esclerodactilia, constricciones digitales (pseudoainhum) y contracturas articulares, con un gran impacto en la calidad de vida de quienes la padecen1–6.
Fue descrita por primera vez en la isla de Mljet (Meleda), Croacia, donde el aislamiento reproductivo y la consanguinidad favorecieron la transmisión y el incremento en la incidencia de esta enfermedad recesiva. Posteriormente se han descrito casos en el Norte de África y en Oriente Medio, siguiendo las rutas comerciales de la República Medieval de Dubrovnik3. En la actualidad su prevalencia estimada en la población general es de 1/100.000 habitantes2.
El estudio genético demostró que la paciente era portadora homocigota del cambio patogénico c.82delT en el gen SLURP-1, asociado a esta enfermedad1–5. No obstante, con los datos disponibles no podemos descartar que sea doble portadora heterocigota de dicha mutación y de una deleción en el gen SLURP-1. Para confirmar la homocigosis habría que analizar dicha mutación en sus progenitores y determinar si ambos son portadores heterocigotos.
Las opciones terapéuticas incluyen emolientes, queratolíticos tópicos, antibioterapia para tratar las sobreinfecciones, retinoides sistémicos e injertos, siendo los retinoides los únicos con eficacia probada, mejorando el engrosamiento epidérmico y las contracturas, no así el eritema2,4.
El mal de Meleda es una queratodermia palmoplantar rara, pero clínicamente tan característica, que debemos recordar.


