
El tumor de células granulares (TCG), descrito por Abrikossof en 1926, es una neoplasia infrecuente, generalmente benigna, considerada de estirpe nerviosa1. Se manifiesta generalmente como una pápula o nódulo solitario. La descripción de lesiones múltiples cutáneas, como en el caso que presentamos1,2, es un hecho muy infrecuente en nuestro medio3.
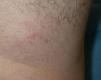
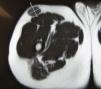
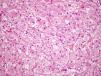
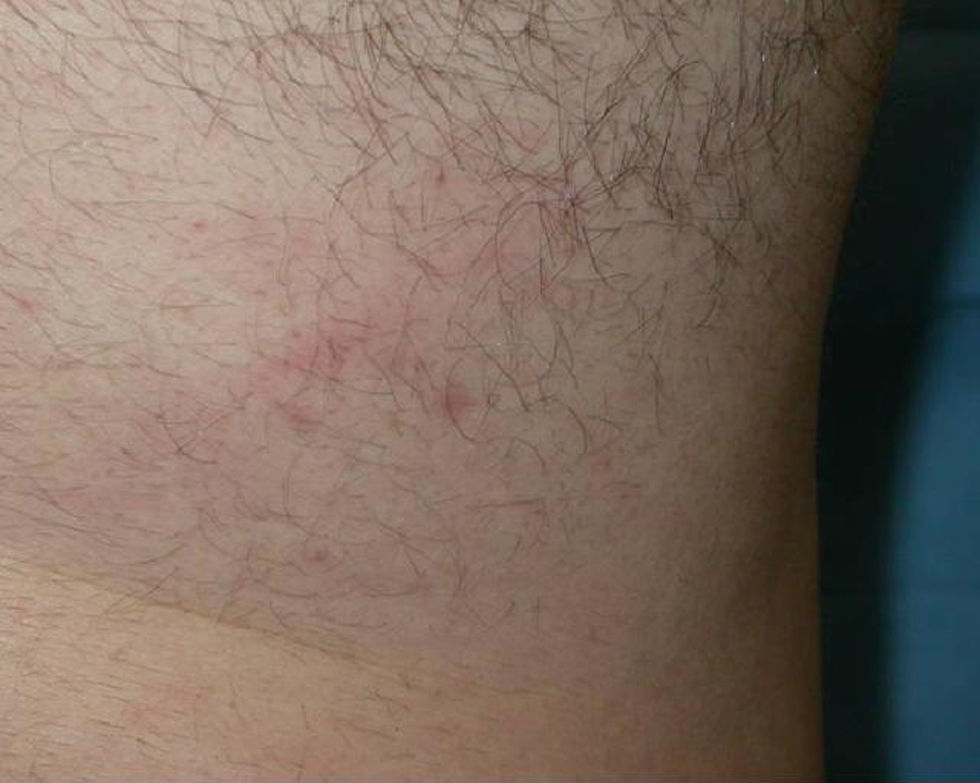
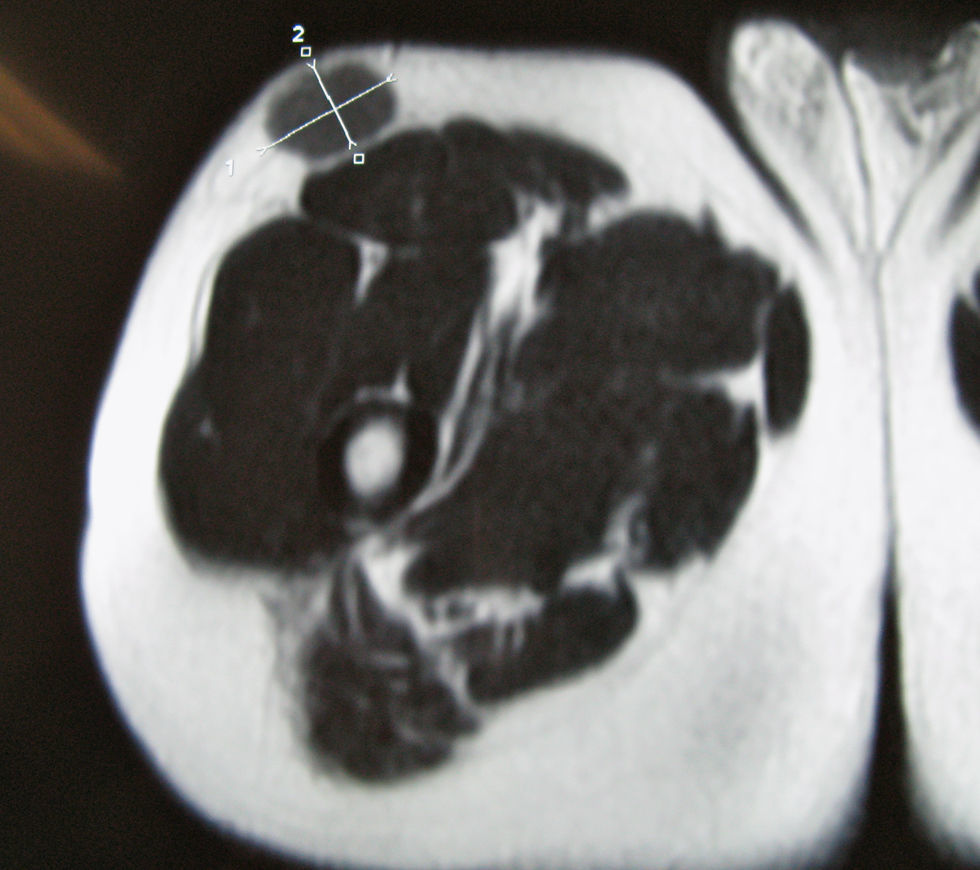
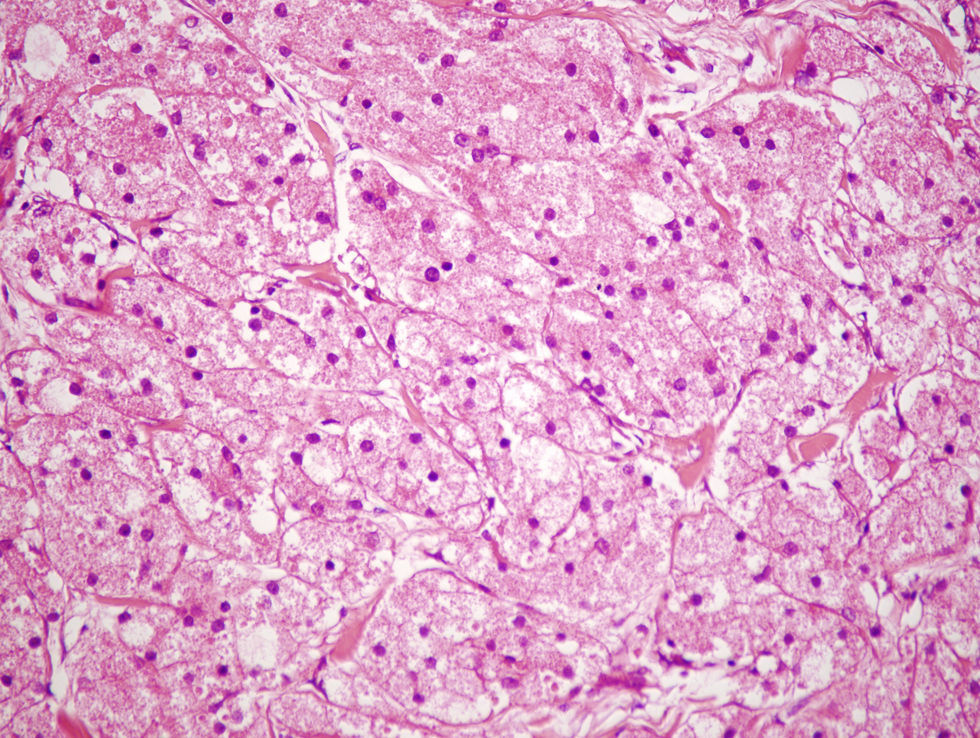
Nuestro paciente es un varón de 41 años de raza blanca, sin antecedentes de interés, que presenta desde hace varios años (3–7 años) 4 tumoraciones subcutáneas de crecimiento lento localizadas en la cadera izquierda, la fosa ilíaca izquierda, el muslo derecho y la región escapular derecha, de entre 1,5 y 4cm (fig. 1), dolorosas a la palpación, no adheridas a los planos profundos. En la resonancia nuclear magnética (RNM) las lesiones se localizaban en el tejido celular subcutáneo y eran independientes de la fascia y el músculo subyacente (fig. 2). La biopsia cutánea mostró una proliferación difusa de células poligonales con citoplasma abundante, granular y eosinófilo, sin necrosis ni mitosis, S-100, vimentina y CD 68 positivas (fig. 3) lo que permitió concluir el diagnóstico de tumor de células granulares para todas las lesiones. Se realizó extirpación amplia de las lesiones por parte del servicio de cirugía plástica. Tras 19 meses de seguimiento no hay signos de recurrencia, enfermedad metastásica, ni han aparecido nuevas lesiones.
El TCG es un tumor infrecuente que supone el 0,017–0,025% de las piezas quirúrgicas2. En series anglosajonas afecta más frecuentemente a mujeres adultas (30–60 años) de raza negra, pero en una serie española predomina en hombres entre la segunda y la quinta décadas3. Su localización más habitual es la lengua (40% de los casos), seguido de la piel y el tejido celular subcutáneo de tórax y extremidades, aunque ha sido descrito en multitud de órganos1,2,4, Se han publicado casos congénitos5 y familiares6. En el 4,5–13% se ha descrito asociado a neoplasias de otra naturaleza1,6,7.
Desde un punto de vista clínico consiste en un nódulo firme, habitualmente menor de 3cm y solitario, circunscrito y asintomático, aunque puede ser pruriginoso o doloroso. El diagnóstico no suele sospecharse clínicamente1–4 y depende del estudio histológico.
En el estudio histológico se caracteriza por ser un tumor mal delimitado no encapsulado formado por sábanas, nidos o cordones de células redondeadas o poligonales de núcleo pequeño y central, y citoplasma amplio y eosinófilo repleto de granulaciones toscas PAS+, diastasa resistente, que representan fagolisososmas1,3. La epidermis que lo recubre puede ser normal o mostrar hiperplasia pseudoepiteliomatosa1–4.
Las técnicas inmunohistoquímicas permiten comprobar que las células granulares son positivas para S-100 (98–100%), ENA (enolasa neuronal específica) (98–100%) y vimentina (100%), y en menor porcentaje para CD 57 (69%) y CD 68 (65%)8.
El tratamiento de elección consiste en la escisión simple de la totalidad de la lesión con un margen suficiente. No se recomienda la radioterapia ni la quimioterapia1–4.
El pronóstico del TCG es excelente. El índice de recurrencias es del 8%1,6 y se asocia generalmente a márgenes de extirpación insuficientes, aunque no todos los casos con márgenes afectos recurren. Los casos malignos (1–2%) se comportan como un sarcoma de alto grado8 y el diagnóstico de malignidad se basa en el comportamiento clínico del tumor, aunque se ha intentado relacionar con aspectos histológicos como la necrosis, el índice mitótico, pleomorfismo, atipia y ulceración8. Son factores pronósticos clínicos adversos: la recurrencia local, la presencia de metástasis, la edad elevada y el tamaño >4cm8.
Los casos múltiples, como el nuestro, suponen, según las series, entre un 0 y un 30% de los pacientes1–6. Esta sorprendente disparidad se ha relacionado con el diferente tiempo de seguimiento, aunque es probable que sea explicado también por diferencias raciales, ya que las lesiones múltiples son más frecuentes en pacientes de raza negra2,6.
Los casos múltiples se asocian en casi el 50% de los niños con alteraciones sistémicas: pigmentarias, cardiovasculares, musculoesqueléticas, del sistema nervioso central y con la neurofibromatosis9.
Por el contrario, en adultos, como es nuestro caso, los TCG múltiples no se han asociado de forma consistente con afectación visceral, otras anomalías, o malignidad. Sin embargo existen dos puntos que queremos destacar:
Multivisceralidad: se han descrito casos de TCG afectando simultáneamente varios órganos. Más concretamente se han descrito casos de TCG múltiples de piel asociados a un TCG gástrico10 y a TCG de pulmón6.
Malignización: en la serie de Khawsur et al se describen 2 casos malignos en un padre y un hijo de raza negra que surgieron en el contexto de lesiones cutáneas benignas múltiples que habían ido apareciendo durante años6.
En conclusión presentamos un caso de TCG llamativo por su carácter múltiple (excepcional en nuestro medio). A pesar de su carácter benigno, y basándonos en la literatura revisada, es aconsejable en estos pacientes realizar una correcta anamnesis por aparatos, una exploración física completa, solicitar pruebas de imagen dirigidas para detectar lesiones viscerales asociadas y hacer un seguimiento periódico.


