


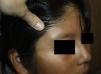
Niña de 12 años sin antecedentes de interés con una lesión en la cola de la ceja derecha de 2 meses de evolución, sin traumatismo previo. Refería dolor ocasional y aumento de tamaño con estabilización posterior.
Exploración físicaTumoración subcutánea ovalada mal definida de 2cm de diámetro, de consistencia elástica firme, adherida a planos profundos y sin alteraciones de la piel suprayacente (fig. 1).
Pruebas complementarias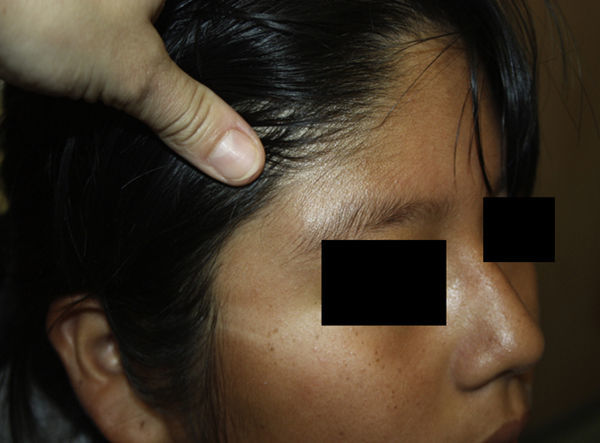
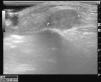
Las radiografías de cara, órbita, hendidura esfenoidal y cráneo fueron normales. La ecografía de partes blandas mostraba una tumoración redondeada y sólida con algún vaso de pequeño calibre en el tejido celular subcutáneo (fig. 2).
Evolución y tratamiento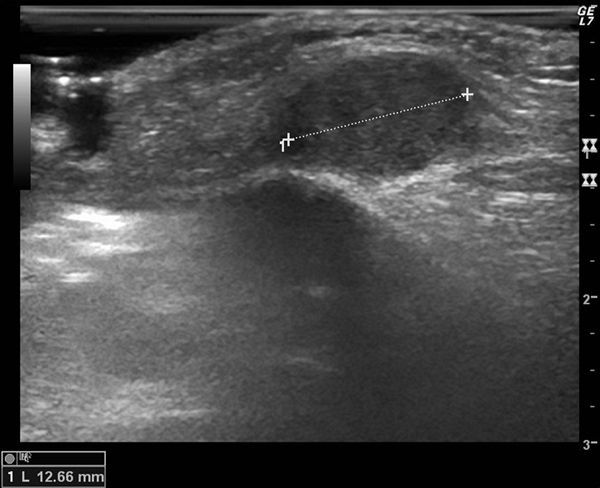
Se procedió a la exéresis completa de la lesión, sin signos de recidiva local ni aparición de otras lesiones tras 8 meses de seguimiento.
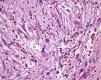
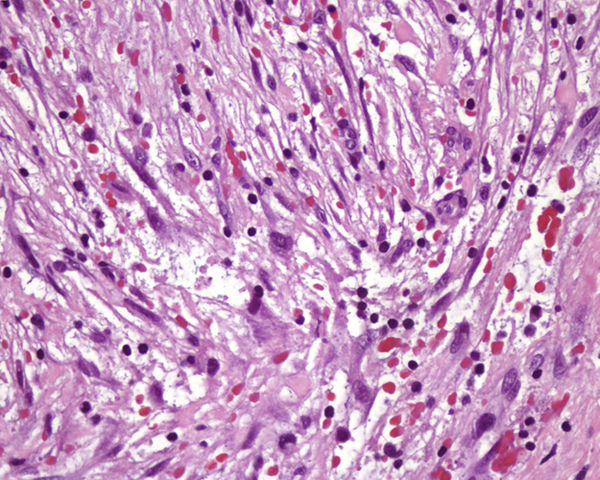
HistopatologíaLa histología mostraba una tumoración localizada en el tejido celular subcutáneo formada por una proliferación de fibroblastos y miofibroblastos fusiformes y multipolares, con núcleo vesiculoso y nucléolo prominente, dispuestos sin un patrón específico en un estroma mixoide con hematíes extravasados y áreas fibrosas con colágeno hialinizado (fig. 3, H-E×400).
¿Cuál es su diagnóstico?
DiagnósticoFascitis nodular (FN).
ComentarioLa FN fue descrita por primera vez por Konwaler et al. en 1954 como «fibromatosis subcutánea pseudosarcomatosa (fascitis)»1. Es una lesión proliferativa de crecimiento rápido que afecta principalmente al tejido celular subcutáneo, la fascia y el músculo, aunque existe una rara variante dérmica2. Su etiopatogenia es incierta; se considera un proceso reactivo, benigno y de crecimiento autolimitado2–4, relacionado en casos aislados con procesos infecciosos o traumatismos2. Predomina en varones de raza blanca3, entre la tercera y cuarta décadas de la vida2; solo un 10% de los casos son niños desde pocos meses de vida hasta los 18 años, con medias de edad según las series entre 8 y 14 años3–5. Se localiza principalmente en las extremidades y el tronco, seguido de la cabeza y el cuello, localización que predomina en la infancia4,5, aunque la casuística infantil es escasa3,5. Suelen presentarse como lesiones únicas y asintomáticas. Clínicamente es un nódulo de consistencia firme, fijo o móvil, generalmente menor de 3cm, de color piel normal o eritematoso, a veces con dolor asociado y sin adenopatías palpables1,3,4.
El diagnóstico es histopatológico. Los estudios de imagen previos ayudan a definir mejor la lesión y descartar otros posibles diagnósticos4.
En el estudio histológico se observa una proliferación de fibroblastos y miofibroblastos con aumento de la actividad mitótica sobre un estroma mixoide laxo2,3, con aumento de la vascularización, eritrocitos extravasados, infiltrado inflamatorio crónico2,4 y en ocasiones células gigantes tipo osteoclasto. Price et al. fueron los primeros en clasificarlas en 3 subtipos histológicos: el tipo i, mixoide o reactivo (el más frecuente), el tipo ii o celular y el tipo iii o fibroso. Se ha propuesto que estos subtipos no son independientes, sino que responden a una progresión temporal2. El estudio inmunohistoquímico muestra positividad para actina muscular específica, actina de músculo liso, calponina y vimentina, y negatividad para desmina, citoqueratina, h-caldesmon, miosina de músculo liso, proteína S100 y CD343,5,6. En estudios citogenéticos se han descrito, como en otros tumores mesenquimales, reordenamientos del locus USP6 relacionados con la alta actividad mitótica4.
El diagnóstico diferencial clínico se realiza con lesiones benignas como el quiste dermoide o epidérmico, el pilomatricoma, el lipoma o el nódulo postoperatorio o postraumático; además de con tumores malignos1–5 como fibrosarcomas, tumor maligno de la vaina nerviosa periférica, sarcoma pleomorfo indiferenciado o dermatofibrosarcoma protuberans.
A pesar de su carácter reactivo y la posibilidad de remisión espontánea, la escisión quirúrgica parece el manejo más razonable, especialmente en la población pediátrica5. Las recidivas son infrecuentes y obligan a replantear el diagnóstico2–5.


