



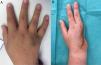

Una niña de 12 años, sin antecedentes, fue derivada a Cirugía Plástica Infantil por presentar una lesión en el dorso de la mano derecha de 3 meses de evolución, consistente en una placa marronácea ligeramente sobreelevada, indurada y dolorosa a la palpación (fig. 1).
Como primera prueba diagnóstica se realizó una ecografía Doppler, objetivándose una lesión subcutánea compatible con malformación arteriovenosa. La resonancia magnética (RM) realizada posteriormente apoyó dicha sospecha. Tras presentar el caso en el Comité de Anomalías Vasculares de nuestro centro, se optó por tratamiento con embolización y posterior cirugía. La embolización no fue posible por imagen compatible con trombosis del nidus malformativo. Al encontrarse asintomática en ese momento, se decidió mantener una actitud expectante con controles ecográficos.
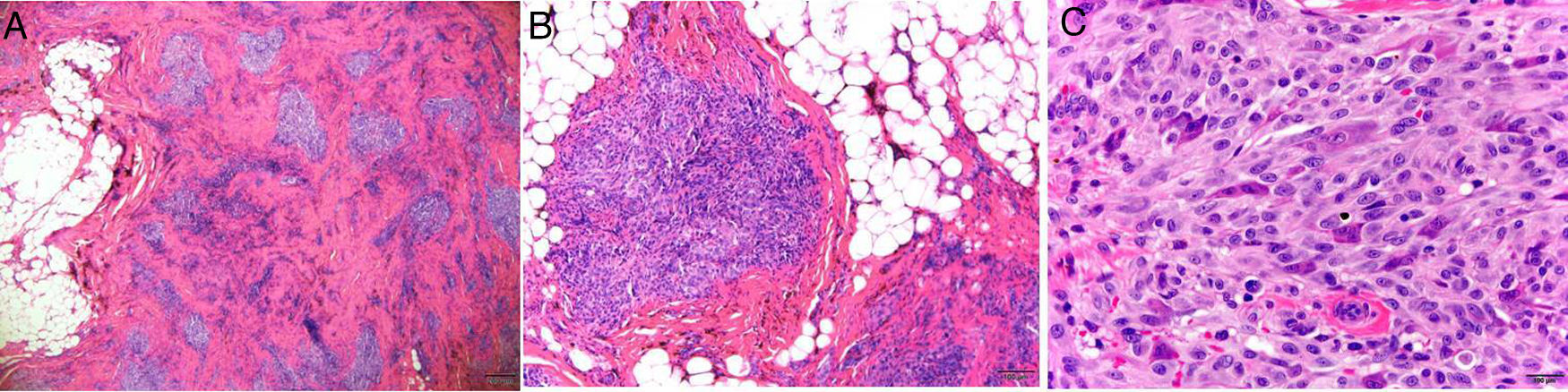
Un año más tarde la paciente solicitó tratamiento por dolor vinculado a traumatismos y la lesión visible fue resecada quirúrgicamente. El estudio histopatológico de la pieza identificó un tumor fibrohistiocitario plexiforme sin márgenes libres. La neoformación afectaba al tejido celular subcutáneo y a las zonas superficiales del músculo esquelético, respetando la piel suprayacente. Presentaba una distribución plexiforme, en nidos tumorales configurados por células fusiformes o epitelioides monomorfas acompañadas de células multinucleadas gigantes. Entre los nidos se advertía un estroma fibroso con vasos dilatados. El índice proliferativo era inferior al 3% y no se identificaban mitosis atípicas, necrosis ni células xantomatosas (fig. 2). El estudio inmunohistoquímico fue negativo para CD31, CD34, WT1 y GLUT1.
Como parte del estudio de extensión, se realizó una tomografía computarizada de tórax en la que se evidenció un nódulo de 3mm en lóbulo inferior derecho. El equipo de Cirugía Torácica Infantil procedió a la resección toracoscópica del mismo, resultando compatible con tumorlet carcinoide.
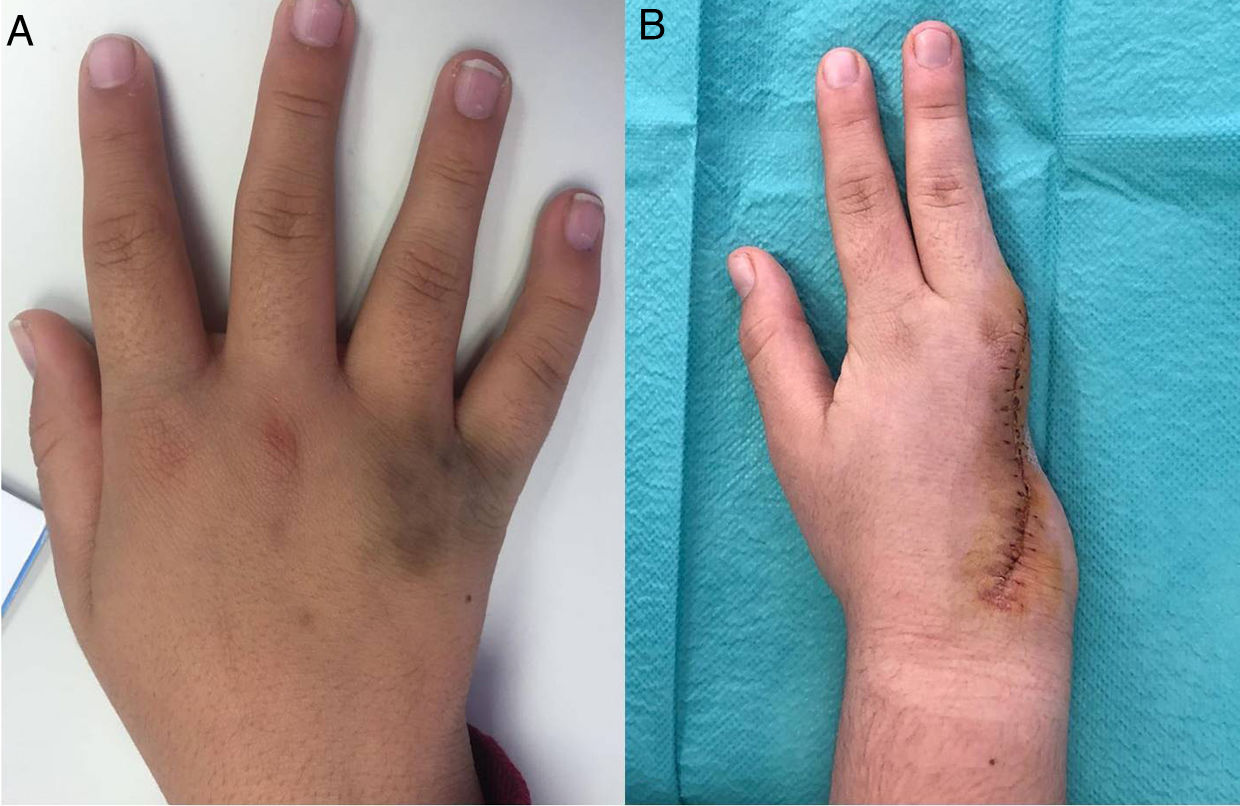
Por otra parte, las pruebas de imagen de la mano derecha demostraron una lesión residual con invasión musculotendinosa y erosión de la cortical del cuarto metacarpiano. Tras discutir el caso en una sesión multidisciplinar compuesta por oncólogos, cirujanos plásticos, traumatólogos, radiólogos y anatomopatólogos, se establecieron 2opciones terapéuticas: observación clínica estrecha o resección completa de la lesión. Finalmente, la familia optó por la cirugía, realizándose una amputación de los dedos 4.° y 5.° de la mano derecha. Actualmente, la paciente mantiene seguimiento, encontrándose asintomática y en proceso de rehabilitación.
El tumor fibrohistiocitario plexiforme, reportado por primera vez por Enzinger y Zhang en 19881, es un tumor de tejidos blandos poco frecuente, con potencial intermedio de malignidad2. Muestra predilección por niños y adultos jóvenes, afectando a menores de 20 años en el 60-70% de los casos3,4. Es más prevalente en el sexo femenino y no predomina en ninguna etnia5. Su etiología es desconocida, pero se han informado casos congénitos y relacionados con traumatismos previos3-6.
Clínicamente aparece como una masa o placa indurada de 1-3cm, indolora y de crecimiento lento que afecta a la dermis y la hipodermis. En ocasiones, produce dolor, ulceración o cambios en la coloración de la piel suprayacente e, infrecuentemente, se extiende al músculo esquelético, como ocurre en el caso reportado por los autores. Suele localizarse en miembros superiores, fundamentalmente dedos, mano o muñeca. También se han reportado casos en extremidades inferiores, tronco, cabeza y cuello3-6.
El diagnóstico inicial supone un desafío desde el punto de vista clínico y radiológico. En la RM se manifiesta como una lesión infiltrativa o en forma de placa centrada en los tejidos subcutáneos, sin características de señal exclusivas que permitan diferenciarlo de otras entidades benignas (entre ellas algunos tipos de anomalías vasculares) o malignas7. La anatomía patológica proporciona el diagnóstico definitivo. Histológicamente, se compone de nódulos o fascículos de histiocitos, fibroblastos y células similares a osteoclastos, dispuestos en un patrón plexiforme característico3. Inmunohistoquímicamente, las células histiocíticas son positivas para CD68 y los fibroblastos expresan actina del músculo liso y vimentina3-6,8.
El tratamiento de elección es la resección quirúrgica con márgenes negativos. La recurrencia local se ha documentado en el 12,5-40% de los casos4, detectándose frecuentemente en los 1-2 años posteriores a la cirugía3. Tras la resección completa, el curso clínico de este tumor es típicamente benigno; sin embargo, se han informado metástasis en los ganglios linfáticos regionales (6%) y el pulmón (2-19%)8. Este hecho hace necesario el seguimiento a largo plazo de los pacientes afectados3-5.
En resumen, describimos un caso de tumor fibrohistiocitario plexiforme en una niña de 12 años en la que la decisión de completar la resección tumoral ante el riesgo, relativamente bajo, de metástasis nodales y pulmonares supuso un conflicto para los médicos tratantes al implicar la amputación de 2dedos de la mano afectada. Finalmente, la familia, quizás influida por el diagnóstico incidental de un nódulo pulmonar en el estudio de extensión, se decantó por la cirugía para evitar el riesgo teórico de metástasis futuras.
Es necesario recalcar la importancia de la anatomía patológica en el diagnóstico de los tumores de partes blandas, existiendo múltiples entidades cuyo manejo y pronóstico pueden diferir significativamente.
Al Dr. Rodríguez y a la Dra. Espinola, patólogos del Hospital 12 de Octubre, de Madrid, por facilitar las imágenes histológicas incluidas en el caso clínico. A los servicios de Traumatología Infantil (Dr. Martí), Cirugía Torácica Infantil (Dra. Morante, Dra. López y Dr. Antón-Pacheco), Hemato-Oncología Pediátrica (Dra. Perez y Dra. Baro) y Radiodiagnóstico Infantil (Dra. Gallego) del Hospital 12 de Octubre, de Madrid, por su contribución en la toma de decisiones y manejo de la paciente.


