


A 31-year-old woman from Cuba was assessed for facial skin lesions that had appeared recently during her admission to the internal medicine department for a study of microcytic anemia, thrombopenia, and kidney failure with proteinuria. The patient had been diagnosed with primary antiphospholipid syndrome (APS) at the age of 13 years after debuting with deep vein thrombosis (DVT) in the right lower limb; since that time, she had not met any of the criteria for systemic lupus erythematosus (SLE). After withdrawal of anticoagulation medication for unknown reasons, prior to her arrival in Spain, the patient experienced new episodes of DVT in both lower limbs, and pulmonary thromboembolisms that caused pulmonary hypertension, requiring pulmonary endarterectomy. Family history included a sister with SLE. The patient was in follow-up with the internal medicine department and had been treated with systemic corticosteroids, cyclophosphamide, azathioprine, mofetil mycophenolate, and rituximab, which achieved partial control of her disease.
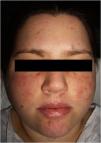
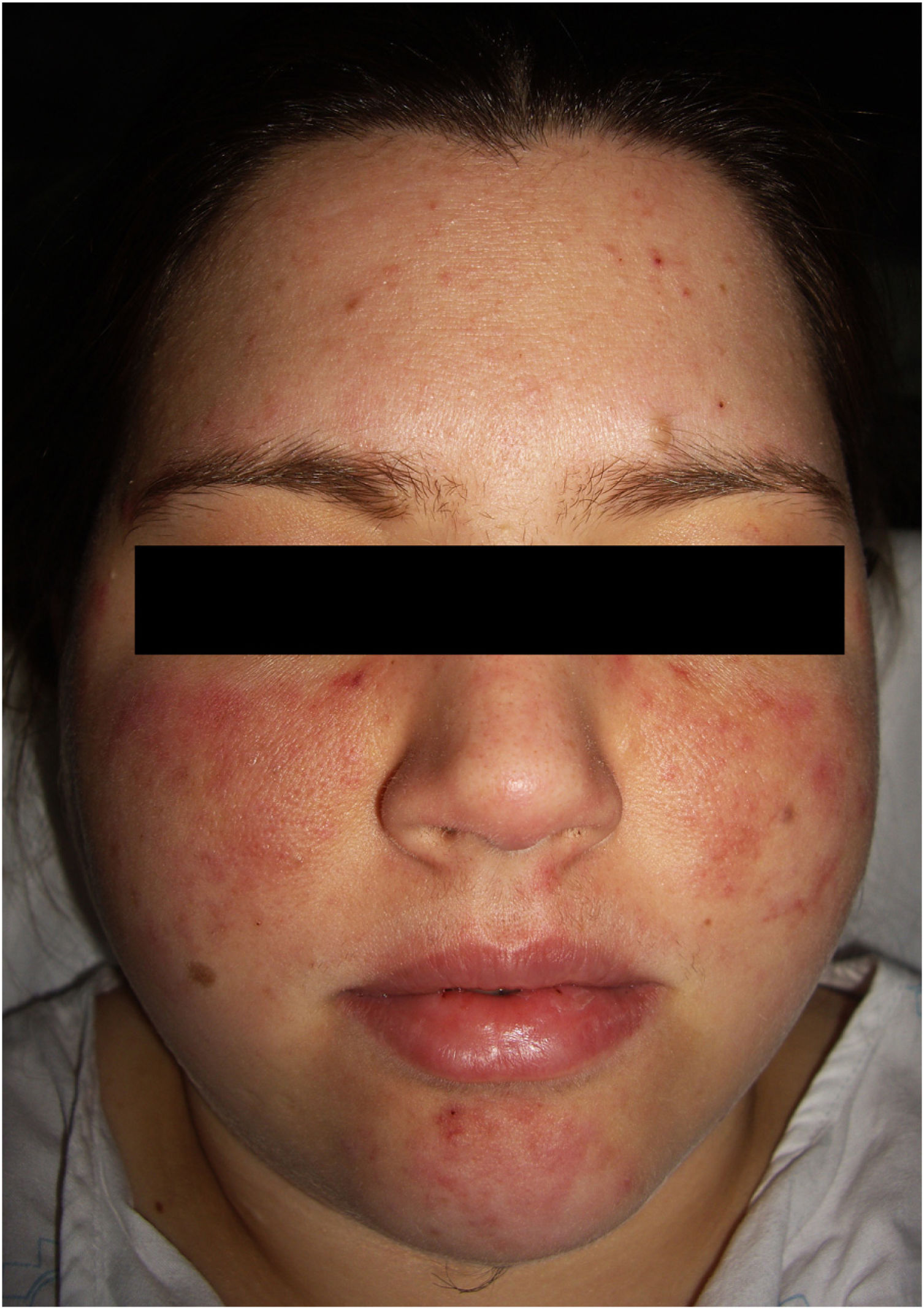
The physical examination revealed 2 symmetrical, erythematous malar plaques, formed by the coalescence of individual papular elements on a background of telangiectasia (Fig. 1). The patient presented similar lesions on the chin and, in a disperse form, on the upper limbs.
Analyses revealed repeatedly negative antinuclear antibodies (ANA), whereas anticardiolipin IgG and anti-beta2-glycoprotein IgG were persistently positive. The rest of the autoimmune study was negative.
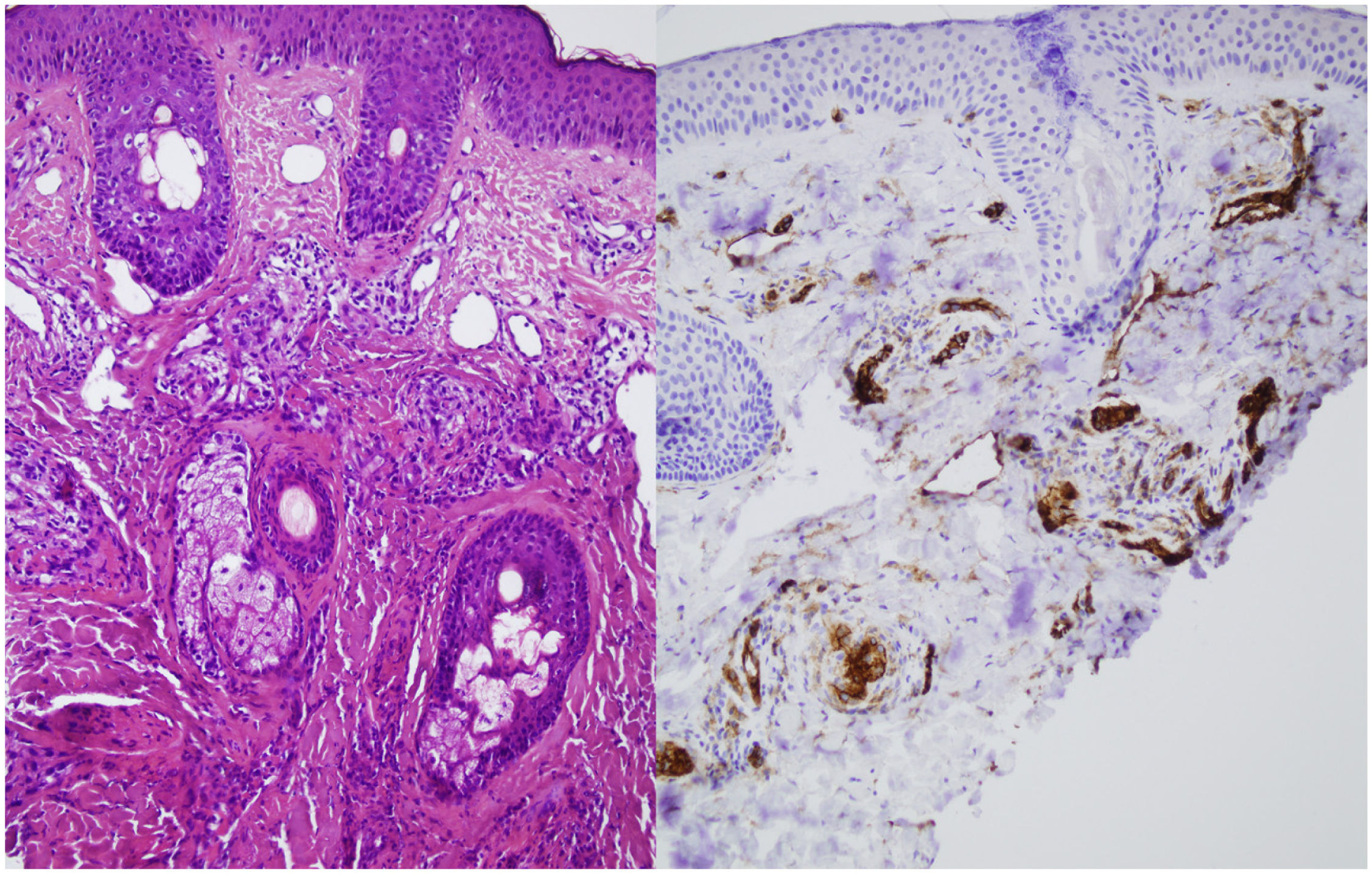
A skin biopsy was performed on one of the malar lesions and revealed dilated capillaries and venules in the superficial dermis, occasionally occupied by a proliferation of fusiform cells without atypia and with clear cytoplasms. No significant inflammatory infiltrates, bleeding or involvement of the interface. Immune staining showed these cells to be endothelial, predominantly positive for CD31 and negative for D2-40 (Fig. 2). This led to a diagnosis of reactive angioendotheliomatosis in a context of primary antiphospholipid syndrome.
Administration of boluses of systemic corticosteroids in combination with immunoglobulins allowed for progressive recovery of the platelet count and partial improvement of kidney function. Treatment was supplemented with the addition of mofetil mycophenolate. The patient is currently maintaining treatment with mofetil mycophenolate, anticoagulation, and low-dose corticosteroids, with a good response of the systemic disease and the skin manifestations.
Angioendotheliomatosis is a cutaneous vascular proliferation of endothelial cells inside the blood vessels. Its reactive form involves a benign proliferation of these cells, usually in the context of systemic diseases involving vascular disease or vascular occlusion.1 In the reactive form, it involves the skin exclusively and is a benign and frequently self-limiting process.1 Its pathogenesis is still unknown. Its association with multiple processes (endocarditis, kidney disease, primary APS and secondary to SLE, valvular heart disease, cryoglobulinemia, rheumatoid arthritis, myelodysplastic syndrome, leukocytoclastic vasculitis) would appear to indicate that it is a common response pattern to various stimuli. All of them share a common mechanism: vascular damage and occlusion, local hypoxia, release of proangiogenic factors (VEGF), and hyperplasia of endothelial cells.1–3
Clinical manifestations include erythematous or purplish macules and plaques. The most common location is on the limbs, although it has also been reported on the torso, head, and neck. The lesions may mimic those found in Kaposi sarcoma, morphea, and scleroderma, pyoderma gangrenosum, angiosarcoma, verrucous lichen planus, sarcoidosis, pyogenic granuloma, and lupus panniculitis.1,2 When it appears on the face, as with our patient, it may mimic a butterfly or malar rash characteristic of SLE.
Histopathology reveals poorly circumscribed, dilated grouped capillaries with endothelial cells without atypia in the dermis and the subcutaneous cellular tissue. The architecture of the CD31+ intravascular proliferation may take on a lobular or diffuse glomerular pattern. These structures are negative for histiocyte markers (CD68) and lymphatic markers (podoplanin or D2-40). Fibrin thrombi or microthrombi are frequently found. The presence of an accompanying lymphocytic infiltrate varies.2,3 The histiologic differential diagnosis includes malignant angioendotheliomatosis and intravascular lymphoma. Histology and immune staining are key to diagnosing this entity.
The treatment of choice is that of the underlying disease, which, in the case of APS, is anticoagulation therapy.1 Cases of reactive angioendotheliomatosis have been reported in patients with APS secondary to SLE and with primary APS,2–4 although the fact that they are already undergoing anticoagulant therapy is not mutually exclusive.3 The atypical presentation of this reactive angioendotheliomatosis mimicking a malar butterfly rash reinforces the need to rule out APS secondary to SLE.
FundingThe authors have not received funding of any kind.
Conflicts of interestThe authors declare that they have no conflicts of interest.


