



Una mujer de 31 años, originaria de Cuba, fue valorada por presentar lesiones cutáneas faciales de reciente aparición durante su ingreso en Medicina Interna en el contexto del estudio de una anemia microcítica, trombopenia e insuficiencia renal con proteinuria. La paciente estaba diagnosticada de síndrome antifosfolípido (SAF) primario desde los 13 años tras haber debutado con una trombosis venosa profunda (TVP) en el miembro inferior derecho, sin cumplir desde entonces en ningún momento criterios de lupus eritematoso sistémico (LES). Tras la retirada de la anticoagulación previa a su llegada a España por motivos desconocidos, sufrió nuevos episodios de TVP en ambos miembros inferiores, así como tromboembolismos pulmonares que condicionaron una hipertensión pulmonar, por lo que precisó una endarterectomía pulmonar. Entre los antecedentes familiares destacaba una hermana con LES. La paciente estaba en seguimiento por el Servicio de Medicina Interna y había sido tratada con corticoides sistémicos, ciclofosfamida, azatioprina, micofenolato de mofetilo y rituximab, con un control parcial de su enfermedad.
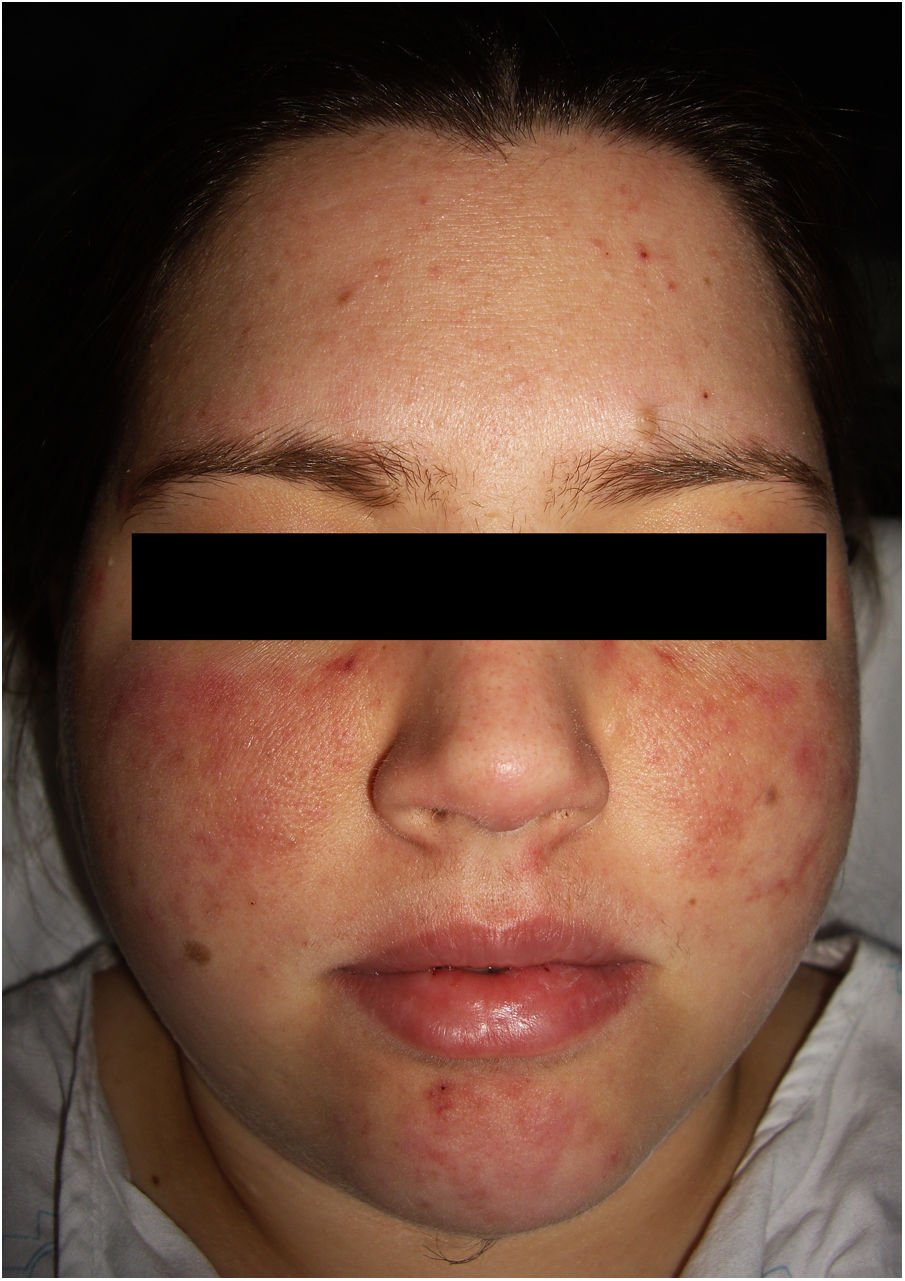
En la exploración física se objetivaron dos placas eritematosas malares, simétricas, formadas por la confluencia de elementos papulosos individuales sobre un fondo telangiectásico (fig. 1). Presentaba unas lesiones similares en el mentón y, de forma dispersa, en los miembros superiores.
La analítica mostraba anticuerpos antinucleares (ANA) repetidamente negativos, mientras que los anticardiolipina IgG y anti-B2-glicoproteína IgG persistían positivos. El resto del estudio de autoinmunidad fue negativo.
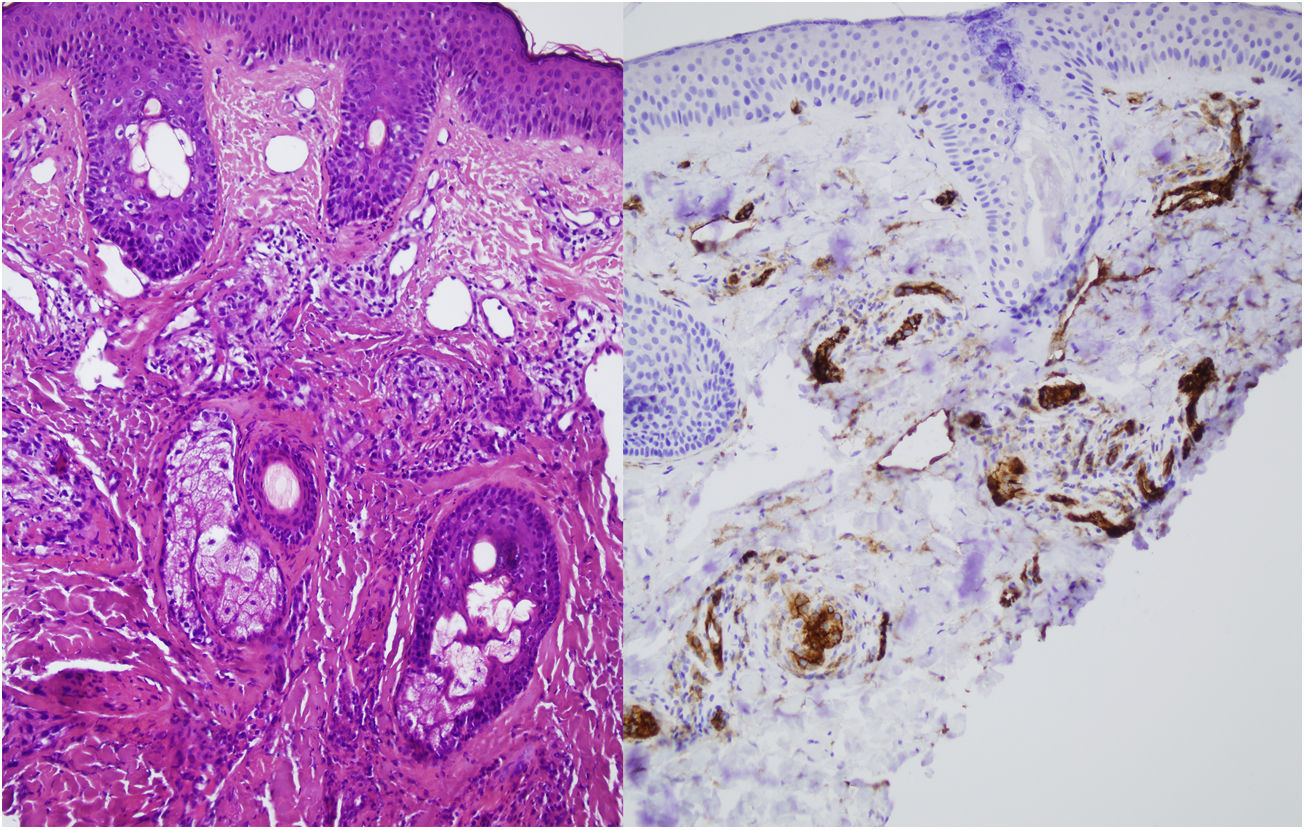
Se realizó una biopsia cutánea de una de las lesiones malares, en la que se evidenció, en la dermis superficial y media, la presencia de unos vasos capilares y venulares de luces dilatadas, ocupados de forma ocasional por una proliferación de elementos celulares fusiformes sin atipia y con citoplasmas claros. No se objetivaron infiltrados inflamatorios significativos, hemorragias ni afectación de la interfase. El estudio inmunohistoquímico demostró que estas células intravasculares eran de naturaleza endotelial, con un predominio de la positividad para CD31 y negatividad para D2-40 (fig. 2). Todo ello permitió el diagnóstico de angioendoteliomatosis reactiva en el contexto de un síndrome antifosfolípido primario.
La administración de bolos de corticoide sistémico junto con la administración de inmunoglobulinas permitió la recuperación progresiva de las cifras de plaquetas y una mejoría parcial de la función renal. El tratamiento se complementó con la adición de micofenolato de mofetilo. Actualmente, la paciente mantiene como tratamiento este último, anticoagulación y dosis bajas de corticoides con una buena respuesta de la enfermedad sistémica y de las manifestaciones cutáneas.
La angioendoteliomatosis es una proliferación vascular cutánea de células endoteliales dentro de los vasos sanguíneos. Su forma reactiva supone una proliferación benigna de la mismas, normalmente en el contexto de enfermedades sistémicas que implican una vasculopatía u oclusión vascular1. En su forma reactiva, tiene implicación exclusivamente cutánea y es un proceso benigno y frecuentemente autolimitado1. Su patogenia todavía es desconocida. El hecho de haberse asociado a múltiples procesos (endocarditis, nefropatías, SAF primario y secundario a LES, valvulopatías, crioglobulinemia, artritis reumatoide, síndrome mielodisplásico, vasculitis leucocitoclástica) parece indicar que se trata de un patrón de respuesta común a varios estímulos. Todos comparten un mecanismo común: el daño y la oclusión vascular, la hipoxia local, la liberación de factores pro-angiogénicos (VEGF) y la hiperplasia de células endoteliales1–3.
La clínica incluye máculas y placas, eritematosas o equimóticas. La localización más frecuente es en los miembros, aunque se han descritos también en el tronco, la cabeza y el cuello. Las lesiones pueden imitar las que se encuentran en el sarcoma de Kaposi, la morfea y la esclerodermia, el pioderma gangrenoso, el angiosarcoma, el liquen plano verrucoso, la sarcoidosis, el granuloma piogénico y la paniculitis lúpica1,2. Cuando aparece en la región facial, como ocurrió en nuestra paciente, pueden simular un eritema en alas de mariposa tan característico de un LES.
El análisis histopatológico revela capilares agrupados, mal circunscritos, dilatados, con unas células endoteliales sin atipia en la dermis y el tejido celular subcutáneo. La arquitectura de la proliferación intravascular CD31+ puede adoptar un patrón lobular, glomerular o difuso. Estas estructuras son negativas para marcadores histiocitarios (CD68) y linfáticos (podoplanina o D2-40). Es frecuente encontrar trombos o microtrombos de fibrina. Es variable la presencia de un infiltrado linfocítico acompañante2,3. El diagnóstico diferencial histológico se plantea con la angioendoteliomatosis maligna o el linfoma intravascular. El estudio histológico e inmunohistoquímico son clave para diagnosticarlo.
El tratamiento de elección es el de la enfermedad subyacente, siendo en el caso del SAF primario la anticoagulación1. Se han descrito escasos casos de angioendoteliomatosis reactiva en pacientes con SAF secundario a LES, así como SAF primario2–4, aunque no es excluyente que éstos se encuentren ya en tratamiento con terapia anticoagulante3. La presentación atípica de esta angioendoteliomatosis reactiva simulando un eritema malar en alas de mariposa refuerza la necesidad de descartar un SAF secundario a LES.
FinanciaciónLos autores no han recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


