




Filaggrin is a structural protein that is fundamental in the development and maintenance of the skin barrier. The function of filaggrin and its involvement in various cutaneous and extracutaneous disorders has been the subject of considerable research in recent years. Mutations in FLG, the gene that encodes filaggrin, have been shown to cause ichthyosis vulgaris, increase the risk of atopic dermatitis and other atopic diseases, and exacerbate certain conditions. The present article reviews the current knowledge on the role of filaggrin in the skin barrier, FLG mutations, and the consequences of filaggrin deficiency.
La filagrina es una proteína estructural fundamental para el desarrollo y mantenimiento de la barrera cutánea. En los últimos años se ha llevado a cabo una extensa investigación sobre su función y su implicación en distintos trastornos cutáneos y extracutáneos. Se ha comprobado que las mutaciones en el gen que la codifica, el gen FLG, son la causa de la ictiosis vulgar y confieren un mayor riesgo de desarrollar dermatitis atópica y otras enfermedades atópicas, además de agravar algunas enfermedades. El presente artículo revisa la información existente en cuanto a su papel en la barrera cutánea, así como respecto a las mutaciones en el gen FLG y las consecuencias que conlleva el déficit de filagrina.
The main function of the skin is to act as a barrier that separates the internal environment from the external one, thus protecting against aggression by exogenous agents and minimizing the loss of water and other essential body components to the external space.1 Filaggrin is particularly important in the formation of the skin barrier, both for its fundamental role in terminal epidermal differentiation and for its implication in some of the most common dermatological diseases, such as atopic dermatitis (AD) and ichthyosis vulgaris.2 Filaggrin is an important structural protein that was first identified in 1977.3 Later, when it was found to produce aggregation and compaction of keratin intermediate filaments, it was named filaggrin, the acronym of filament-aggregating protein.4 This protein is synthesized as a giant precursor protein called profilaggrin, which is the main component of the keratohyalin granules in the stratum granulosum of the epidermis.
Role of Filaggrin in the Formation of the Epidermal BarrierThe main element of the skin barrier is the stratum corneum. This stratum is the end-product of the differentiation of keratinocytes, which, from the basal layer to the granulosum layer, are viable nucleated cells. These cells express various structural proteins as they mature.5 In the final steps of differentiation, the keratinocytes undergo marked changes in their structure, leading to their transformation into flat, anucleate squamous cells, the corneocytes. These corneocytes, which remain tightly bound together by corneodesmosomes, are covered by a cellular coating called the cornified envelope (CE), which has protein and lipid components that endow the cells with mechanical and chemical resistance.5 Between the cells there is a hydrophobic, lipid-rich extracellular matrix arranged in a laminar bilayer.6 This organization of the stratum corneum has been called “bricks and mortar”, in which the keratinocytes are the bricks and the extracellular lipid matrix is the mortar.7
A calcium gradient exists in the epidermis, with low concentrations in the basal layer, even lower concentrations in the stratum spinosum, high levels in the stratum granulosum, and very low levels in the stratum corneum.8 This gradient is important in terminal keratinocyte differentiation.2,9 The higher concentration of calcium in the stratum granulosum causes the keratohyalin granules to release their contents, leaving the profilaggrin exposed to undergo processing and fragmentation into active filaggrin monomers.10 This free filaggrin binds to the intermediate keratin filaments, causing their aggregation and compaction, provoking the collapse and flattening of the cell. Simultaneously, the cell expresses a series of structural proteins that make up the protein portion of the CE.11,12 The bundles of keratin intermediate filaments aggregated by filaggrin bind to these structural proteins through the action of transglutaminases.6 In addition, the increase in the calcium concentration also provokes release of the contents of the lamellar bodies, which are granules rich in lipids and enzymes synthesized in the Golgi apparatus. The action of these enzymes on the lipids gives rise to the lipid portion of the CE and the extracellular matrix of the stratum corneum.6,11,13
Filaggrin continues to undergo processing by various proteases. This proteolysis leads to the release of hygroscopic amino acids and their derivatives, which form natural moisturizing factor (NMF), responsible for water retention in the stratum corneum.14 The breakdown of some of these amino acids gives rise to 2 organic acids: trans-urocanic acid (UCA), a histidine derivative; and pyrrolidone-5-carboxylic acid (PCA), a glutamine derivative. These acids are 2 of the main factors responsible for maintaining the acid pH of the stratum corneum,15 which is essential for its role in the metabolism and organization of the lipids of the extracellular matrix,16 for its antimicrobial action, and for its regulatory role on enzyme activity and physiologic desquamation.17 In addition, UCA has a photoprotective effect against UV radiation18 and has been shown to be a key mediator in UV-B–induced immunosuppression.19,20
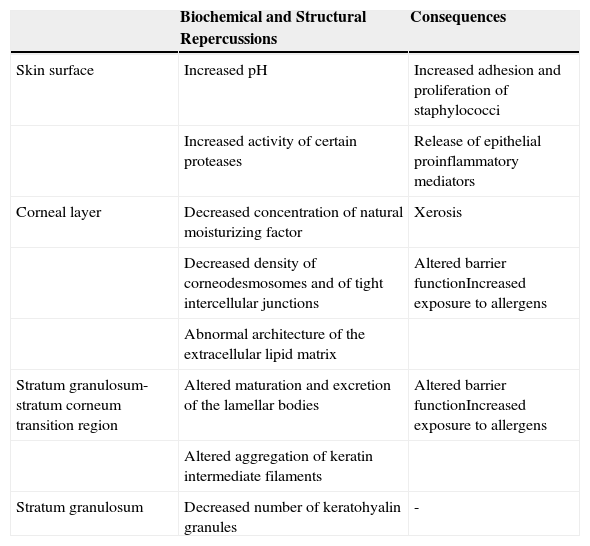
The Epidermal Barrier and Filaggrin DeficitFilaggrin deficit has a major impact on the epidermal barrier (Table 1), affecting the organization of the keratin filaments of the cytoskeleton and the structure of the CE. There is also a fall in the number of keratohyalin granules, a marked fall in NMF concentration (and thus in hydration of the stratum corneum), and alkalinization of the skin pH.5 In addition to these changes, recent ultrastructural studies have shown that a deficit of filaggrin is also associated with a generalized fall in the density of corneodesmosomes and of tight intercellular junctions, as well as with abnormalities in the architecture of the extracellular lipid matrix; these changes may produce a notable alteration of barrier function, provoked by abnormalities in the organization of the cytoskeleton (which affect lamellar body maturation and exocytosis, leading to a nonuniform distribution of secreted lipids and enzymes) and by the increase in the pH (which modulates the activity of those enzymes).16 Furthermore, the increase in the activity of certain proteases caused by the persistent elevation of the pH favors the release of proinflammatory mediators by keratinocytes; these mediators induce an inflammatory response mediated by type 2 helper T (Th2) cells even in the absence of allergens.21 For example, alkalinization of the skin pH increases the activity of the proteases responsible for the production of interleukin (IL) 1α and IL-1β from their inactive proproteins generated by keratinocytes.22
Effects of Filaggrin Deficit on the Epidermal Barrier.
| Biochemical and Structural Repercussions | Consequences | |
|---|---|---|
| Skin surface | Increased pH | Increased adhesion and proliferation of staphylococci |
| Increased activity of certain proteases | Release of epithelial proinflammatory mediators | |
| Corneal layer | Decreased concentration of natural moisturizing factor | Xerosis |
| Decreased density of corneodesmosomes and of tight intercellular junctions | Altered barrier functionIncreased exposure to allergens | |
| Abnormal architecture of the extracellular lipid matrix | ||
| Stratum granulosum-stratum corneum transition region | Altered maturation and excretion of the lamellar bodies | Altered barrier functionIncreased exposure to allergens |
| Altered aggregation of keratin intermediate filaments | ||
| Stratum granulosum | Decreased number of keratohyalin granules | - |
Profilaggrin is coded by the FLG gene, located in the epidermal differentiation complex on chromosome 1 (locus 1q21), a cluster of genes that code for proteins involved in epidermal differentiation.23 The FLG gene is made up of 3 exons and 2 introns (Fig. 1). Transcription starts at exon 2, but it is exon 3 that codes for the greatest part of the protein, constituting one of the largest exons in the genome.24 The resulting protein (profilaggrin) is rich in histidine and contains between 10 and 12 repetitions of filaggrin flanked by N- and C-terminus domains (Fig. 1).

These domains are also functional. The N-terminus domain is, in turn, formed by 2 subdomains: A andB. Subdomain A contains 2 calcium binding sites.25 It is the binding of calcium to this subdomain that produces a series of conformational changes in the profilaggrin molecule that will initiate its processing.2 The B subdomain contains a nuclear localization signal that facilitates translocation of the N-terminus domain to the cell nucleus when it is cleaved from the rest of the protein.26 It has been suggested that this N-terminus domain, once within the nucleus, plays an important role in the loss of the keratinocyte nucleus during transformation of the cell into an anucleate corneocyte27 and it has also been attributed a role in assembly of the CE.28 The C-terminus domain is essential for correct processing of the profilaggrin, although its exact function is unknown.2
Mutations of the Filaggrin Gene (FLG)The first mutations identified in the FLG gene were R501X and 2282del4 in patients with ichthyosis vulgaris.29 Many more mutations have been described since that time, but all are loss-of-function mutations (amino acid substitutions or frameshift mutations).21 Furthermore, the mutations have been shown to display ethnic/geographic specificity, with different mutations being detected in the European and Asian populations.30 There is also variation in the frequency of the mutations: 2 mutations (R501X and 2282del4) account for 80% of all FLG mutations in Ireland, whereas in Singapore no single mutation predominates in any such way over the others.21 The overall prevalence of mutations in the general population in Europe is 7.7%, but in Asia is 3%.31 Focusing on European studies, there is a noticeable difference in prevalence between the north and south of the continent. The majority of publications refer to northern populations, with a prevalence of approximately 10% (range, 7%-14%).31 There are only 2 studies in Mediterranean populations, one in a French population32 and the other in an Italian population33; both studies revealed a much lower prevalence (4%).
This apparently latitude-dependent trans-European gradient, led the Italian authors to put forward that perhaps FLG mutations offer some form of survival benefit in the north.33 It has been suggested that this could be related to a lower level of UCA found in these patients; this would produce greater sensitivity to UV radiation.18,32,34 Recent population-based studies have revealed that individuals with FLG mutations have serum vitamin D levels that are 10% higher than controls.35 Overall, these data show that in latitudes with a lower intensity of UV radiation, individuals with FLG mutations may have greater protection against the onset of diseases such as rickets.31
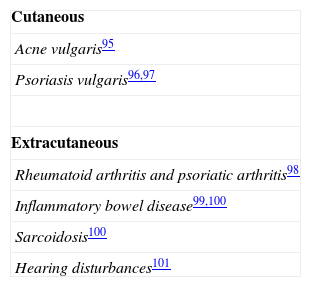
Table 2 lists the disorders that have been associated with a filaggrin deficit caused by mutations in the FLG gene; the main diseases are ichthyosis vulgaris and atopic disorders.
Ichthyosis VulgarisIchthyosis vulgaris is the most common disorder of keratinization, with an estimated prevalence between 1 in 80 and 1 in 250 in school-age English children,15 and is the disease of mendelian inheritance caused by mutations in the FLG gene.31 The absence (or marked reduction) of keratohyalin granules in biopsies from patients with ichthyosis vulgaris was first observed in the 1980s, together with a decrease in filaggrin expression detected using immunostaining techniques.36 However, it was not until 2006 that loss-of-function mutations in the FLG gene were identified as the cause of the condition29; this lag was the result of the long length of the gene and its highly repetitive sequence, which made it difficult to sequence using conventional polymerase chain reaction techniques.15 The discovery of these mutations made it possible to clarify the pattern of disease inheritance; this is autosomal semidominant, which explains the phenotypic variability of ichthyosis vulgaris. Heterozygotic patients present haploinsufficiency, which is a 50% reduction in filaggrin expression. In this situation, patients develop less serious disease (or may even be asymptomatic) and the manifestations respond better to external influences, such as the application of emollient creams or environmental moisture. Homozygotic individuals, on the other hand, develop all the manifestations of the disease.21,29
Clinically, ichthyosis vulgaris is characterized by the appearance of xerosis, keratosis pilaris, palmar hyperlinearity, and atopy in the postnatal period (typically in the first year of life).21 Xerosis is seen as a fine, sometimes polygonal desquamation that mainly affects the extensor surface of the limbs (Fig. 2), scalp, central facial region, and trunk, while the folds are usually spared. The level of moisture modifies filaggrin processing,14,37 which explains the tendency to sparing of the skin folds and why, in the majority of patients, the disease worsens in winter, when environmental humidity is lower.31

Painful fissures on the hands, heels, fingers, and toes are common (present in up to 76% of patients with ichthyosis vulgaris). This is closely related to the level of environmental moisture.31 Furthermore, mutations in the FLG gene have been associated with the onset of fissured dermatitis on the dorsum of the hands and fingers in patients with AD and even in the general population.31,38
Palmar hyperlinearity (Fig. 3) and keratosis pilaris are typical findings not only in patients with clinical manifestations of ichthyosis vulgaris but also in all individuals with FLG mutations. One study found that palmar hyperlinearity had a positive predictive value of 71% and a negative predictive value of 90% for mutations in the FLG gene. That is, 71% of children with palmar hyperlinearity have a mutation, whereas mutations are highly unlikely in patients without palmar hyperlinearity. In the case of keratosis pilaris, the positive predictive value was 53% and the negative predictive value was 90%.39

Finally, 35% to 70% of patients with ichthyosis vulgaris develop atopic disease, particularly dermatitis but also allergic rhinitis and asthma.40 This finding has led to extensive research into the relationship between mutations in the FLG gene and atopic diseases, detecting interesting data on the pathogenesis of these complex disorders.
Atopic Diseases: Atopic MarchAtopy is defined as a personal or familial tendency to sensitization and immunoglobulin (Ig) E antibody production in response to exposure to common environmental allergens to which everyone is exposed but to which the majority develop no response.41 This tendency predisposes to the development of the so-called atopic diseases, which affect 20% of the population in developed countries.42 The concept of atopic march was introduced to describe the tendency of AD to precede the sequential onset of asthma and allergic rhinitis,43 which suggests that AD has an initiator role in the process. It has recently been suggested that food allergies may also form part of this atopic march.44
Mutations in FLG and the Initiation of Atopic MarchIn 2006, the same year that the relationship between ichthyosis vulgaris and mutations in the FLG gene was described, a significant association was reported between the mutations and the onset of AD, the first step of atopic march.45 Traditionally, AD has been considered an immune-mediated disorder associated with a secondary alteration of the skin barrier. However, particularly since the discovery of the role of filaggrin in many patients, the currently predominant thought is that the primary disorder is of the skin barrier.40 According to this new hypothesis on the pathogenesis of AD, all patients with the disease have an inherent defect of the skin barrier, and such a defect has actually been demonstrated in both diseased and disease-free skin.46,47 This defect can be caused by various different molecular mechanisms, one of the most important and frequent of which is filaggrin deficit secondary to mutations in the FLG gene.15 The alteration of the skin barrier, combined with the immunological changes that it induces, will give rise to the clinical manifestations of the disease.21,40,42
Influence of FLG Mutations on the Immune SystemThe alteration of the stratum corneum allows allergens to pass through and be taken up and processed by the Langerhans cells of the epidermis. The Langerhans cells migrate to the lymph nodes where they interact with T cells and induce a Th2-mediated immune response.40,42,48 Percutaneous sensitization through a defective skin barrier has been demonstrated in mice with mutations in FLG49–51 and it has also been shown that patients with AD and mutations in FLG have a significantly higher frequency of allergen-specific Th2 responses.52 The continual entry of allergens will end up causing polarization of the adaptive immunity towards Th2,42 characterized by the local production of Th2 cytokines (IL-4, IL-5, and IL-13), increased eosinophil and mast cell production, survival, and activation, and the production of allergen-specific IgE with an increase in total IgE.40 In addition, this polarization towards Th2 is also favored by a series of cytokines released by the altered keratinocytes,40 such as IL-1 and thymic stromal lymphopoietin (TSLP), as a result of the increased activity of endogenous proteases22 and the action of exogenous proteases (including those released by sources of allergens [e.g., dust mites, roaches, fungi, and pollens] and those produced by Staphylococcus aureus, which frequently colonizes the skin of these patients).53
Mutations in FLG and the Progression of Atopic MarchRecent studies have demonstrated that carriers of FLG mutations have a higher risk of progression of atopic march,54 and have shown a significantly higher risk of developing asthma,55 allergic rhinitis,55 and peanut allergy56 compared with noncarriers.
Filaggrin is not expressed in the bronchial, nasal, or gastrointestinal mucosa and there will therefore be no alteration of barrier function at this level.40,57,58 The mechanism by which FLG mutations promote allergic responses in these mucosas is therefore through systemic sensitization to allergens that have penetrated through a defective skin barrier, rather than through the mucosa. This explains the tendency of AD to precede the other atopic disorders of atopic march.5 A correct treatment of AD in children that restores and maintains the epidermal barrier would therefore prevent the subsequent onset of asthma, allergic rhinitis, or food allergy.42 This supports the growing tendency to maintain a proactive (rather than reactive) therapeutic attitude towards AD, particularly in cases with moderate or severe manifestations.59,60
Atopic DermatitisFLG Mutations: Role in the Risk, Severity, Manifestations, and Epidemiology of Atopic DermatitisAD affects 20% of children and has a marked impact on their quality of life. It is the most prevalent chronic disease of childhood.61 More than 30 studies have confirmed its association with mutations in FLG5 and 2 recent meta-analyses have reported an odds-ratio (OR) for AD in association with FLG mutations of 3.1262 and 4.78,63 respectively. Carriers of FLG mutations therefore have a 4-times higher risk of developing AD than noncarriers. However, there appears to be not only a higher risk, but also greater severity.64 Overall, “only” 15% to 20% of European patients with AD have mutations in FLG.10 However, if European patients with AD are grouped according to clinical severity, it is found that the mutations are much more common in the group with moderate or severe AD (50%) than in those with mild AD (4%-15%).65
Based on the combined data from all the studies performed to date, the clinical profile described for AD associated with mutations in FLG (ADFLG) differs from the profile of those who do not have mutations (ADnon-FLG).5 ADFLG patients have more severe disease,64 an earlier onset,66,67 a greater tendency of the disease to persist into adulthood,68 more marked alkalinization of the pH of the stratum corneum,69 much lower levels of NMF,5 greater IL-1β production in the stratum corneum,22 palmar hyperlinearity,39 more common fissured dermatitis on the dorsum of hands,38 higher serum IgE levels,48 greater allergic sensitization,48 the development of multiple allergies,63 a higher risk of asthma,55,70 and a 10-fold risk of eczema herpeticum.71
In addition, this division also appears to be important from an epidemiological research point of view. The prevalence of AD is much higher in industrialized countries, leading to a suspicion of the influence of environmental factors.5 Recent studies have shown that children who live with cats (but not with dogs) at early ages,72,73 and those with an older sibling (which may imply greater exposure to pathogens and allergens)74 have a significantly higher risk of developing AD. But this risk is only present in those children with mutations in FLG. Similarly, clinical severity correlates with the presence of specific IgE for dust mites or cat dander among patients with ADFLG but not among those with ADnon-FLG.10 It would thus appear important that future epidemiological studies of AD should stratify patients into carriers and noncarriers of mutations in FLG.5
FLG Mutations and S aureus in Atopic DermatitisAccording to some studies, more than 90% of patients with AD are colonized by S aureus (vs 5% of healthy subjects)5,75 and the clinical severity of the dermatitis correlates with the number of S aureus present on the skin.76 This colonization is dependent on the integrity of the skin barrier and on the expression of bacterial adhesion proteins, which also contribute to the inflammatory state.5 It has been shown in vitro that an acid pH of the stratum corneum inhibits the expression of these proteins on the bacterial surface and that the filaggrin degradation products, UCA and PCA, are themselves able to inhibit the expression of some of these proteins independently of the pH.77FLG mutations will thus cause greater S aureus colonization not only by altering the skin barrier but also by increasing the expression of adhesion proteins by the bacteria, through alkalinization of the pH of the stratum corneum and reduced levels of UCA and PCA. In addition, the Th2 environment that exists in the lesions of these patients (and that is due in part to the deficit of filaggrin) inhibits the expression of antimicrobial peptides.78
Furthermore, S aureus secretes a wide variety of virulence factors that exacerbate the inflammation, worsening the skin lesions and interfering with their healing.77 The most important of these virulence factors is α-toxin, which induces cell death by creating pores in the plasma membrane that provoke cytolysis. A recent study has shown in vitro that keratinocytes with filaggrin deficit are more vulnerable to the action of this toxin than keratinocytes with normal filaggrin expression.79 This is because cells that do not express filaggrin also have reduced sphingomyelinase expression, which leads to an increased presence of sphingomyelin, a lipid of the plasma membrane that serves as a receptor for α-toxin.79
In summary, FLG mutations will not only facilitate colonization by S aureus but will also make keratinocytes more vulnerable to the action of S aureus toxins. On this subject, in 1 study it was observed that AD patients with an FLG mutation (compared with noncarriers) had a 7-fold risk of developing more than 4 episodes a year of skin infection that required antibiotic treatment.80
Therapeutically, these data support the need for measures that reduce colonization by S aureus and that acidify the skin pH to impede colonization.77
Other Mechanisms of Filaggrin Deficit in Atopic DermatitisAlthough FLG mutations can explain many cases of AD (particularly the severe cases), a large proportion of these patients do not have any mutation.10 However, it has been observed that the expression of filaggrin is also reduced in these patients without mutations, though to a lesser degree.10 It would thus appear that additional mechanisms are involved in the deficit of filaggrin.
Th2 CytokinesAcute AD lesions have a predominantly Th2 inflammatory response to the entry of external antigens through the altered skin barrier.5,81 It has been shown that in vitro exposure of keratinocytes to Th2 cytokines produces a significant reduction in the expression of filaggrin.82 Many patients with AD but without mutations in FLG may thus have an acquired filaggrin deficit because of the Th2 environment. This would in turn cause greater alteration of the stratum corneum, with increased entry of antigens that would finally lead to greater Th2 immune polarization, creating a vicious circle. To this situation it must be added that Th2 cytokines also cause a reduction in the expression of CE proteins such as loricrin and involucrin, further altering the structure of the stratum corneum.83 Again, this would justify an active therapeutic approach to break these vicious circles.59
Variation in the Number of CopiesAs stated above, profilaggrin coded by the FLG gene contains between 10 and 12 filaggrin monomers.2 This is called copy number variation and a recent study has demonstrated that this has clinical relevance.84 In that study in Ireland it was observed that the most common variant of profilaggrin was the one containing 11 monomers (51.5%), followed by the one with 10 monomers (33.9%) and the one with 12 monomers (14.6%); those authors found that the variation in the number of copies gave rise to a dose-dependent decrease in the risk of AD. A subject whose 2 alleles coded for 10 monomers had a risk of developing AD 1.67 times higher than a patient whose 2 alleles coded for 12 monomers; the risk fell with each additional monomer.84 Furthermore, a lower number of copies also correlated with a lower level of UCA in the stratum corneum.84 Using the data obtained in that study, the authors estimated that an increase of 5% to 10% in filaggrin levels can have clinical repercussions. This would appear to be a good starting point for the development of new treatments.
Epigenetic Regulatory MechanismsEpigenetic mechanisms alter the expression of a gene without changing its nucleotide sequence. At present, the only data on this subject relevant to filaggrin come from a study in which it was observed that, in individuals with haploinsufficiency, the higher the degree of methylation of the FLG gene, the higher the risk of developing AD.85 It is believed that the increased risk of AD is due to a lower expression of the gene caused by the methylation, but that parameter was not evaluated.85
Asthma, Allergic Rhinitis, and Food AllergyForty percent of children with AD will finally develop asthma or allergic rhinitis.10 In the case of asthma, the risk correlates with the severity of the skin disease, with 70% of patients with severe AD developing asthma, while this only occurs in 20% to 30% of those with mild AD and 8% of the general population.42 Population-based studies have demonstrated that carriers of FLG mutations have a higher risk of developing asthma, with an overall OR of 1.8.55,70 But that risk is limited to those with AD, in whom the OR rises to 3.3 (95% confidence interval, 3.16-3.49).55,70 Carriers of a mutation also have a higher risk of allergic rhinitis (OR, 2.64), although in this case it is independent of whether or not the patient has eczema.55 Finally, concerning the onset of food allergy, it has been shown that carriers of FLG mutations have a significantly higher risk of developing peanut allergy; this risk is greater if the patient has AD, but is also present independently of AD (overall OR, 5.3, with a residual OR of 3.8 after adjustment for AD).56
Role of FLG Mutations in Other DisordersSince the discovery of mutations in the FLG gene, numerous studies have been performed to look for a possible relationship with various skin and extracutaneous diseases, whether due to an association with AD, to a similar underlying pathogenesis, or to a shared susceptibility locus.15 A summary of the results of these studies is presented in Tables 2 and 3. Given the high frequency of carriers in the general population, it is likely that FLG mutations can act as a modifying factor in numerous disorders, particularly in those related to abnormalities of keratinization and of the skin barrier.21
Disorders That Have Been Associated With Mutations in FLG.
| Cutaneous |
| Ichthyosis vulgaris29 |
| Atopic dermatitis45and its complications:Recurrent Staphylococcus aureus infection80Eczema herpeticum71 |
| Irritant contact dermatitis86,87 |
| Nickel sensitization and earlier onset allergic contact dermatitis to nickel88,89 |
| Chronic hand eczema90 |
| Alopecia areata: more aggressive course in patients with atopic dermatitis91 |
| More severe phenotype of recessive X-linked ichthyosis92 |
| More severe phenotype of pachyonychia congenita93 |
| Extracutaneous |
| Asthma55,70 |
| Allergic rhinitis55 |
| Peanut allergy56 |
| Type 2 diabetes mellitus94 |
Source: Adapted from Thyssen et al.51
Filaggrin is an essential protein for the correct formation and function of the skin barrier. Mutations in the FLG gene are responsible for ichthyosis vulgaris and are associated with a higher risk of developing AD, asthma, allergic rhinitis, and food allergy. The discovery of its function has enabled us to better understand the pathogenesis of various disorders associated with alterations of the skin barrier, and it is likely that its mutations influence the onset or clinical severity of other dermatologic diseases. In addition, the study of its functions and of the consequences of its deficit may have important therapeutic implications in the future, with the possibility of developing new specific treatments for disorders in which this gene is altered.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Armengot-Carbo M, Hernández-Martín Á, Torrelo A. Filagrina: papel en la barrera cutánea y en el desarrollo de patología. Actas Dermosifiliogr. 2015;106:86–95.


