


A 47-year-old man with a past history of hepatitis C and negative human immunodeficiency virus (HIV) serology presented with an asymptomatic lesion on the left upper eyelid that had first appeared more than 5 years earlier. The lesion began as a “pimple” and grew progressively. The patient reported no exposure to toxic chemical products or to radiotherapy. He had received a diagnosis of chalazion from the ophthalmology department.
Physical ExaminationPhysical examination revealed a single, well-defined, brownish-yellow tumor on the left upper eyelid that was keratotic with an erythematous base, which made contact with the edge of the eyelid (Fig. 1). The patient had no palpable locoregional lymph nodes.
Histopathology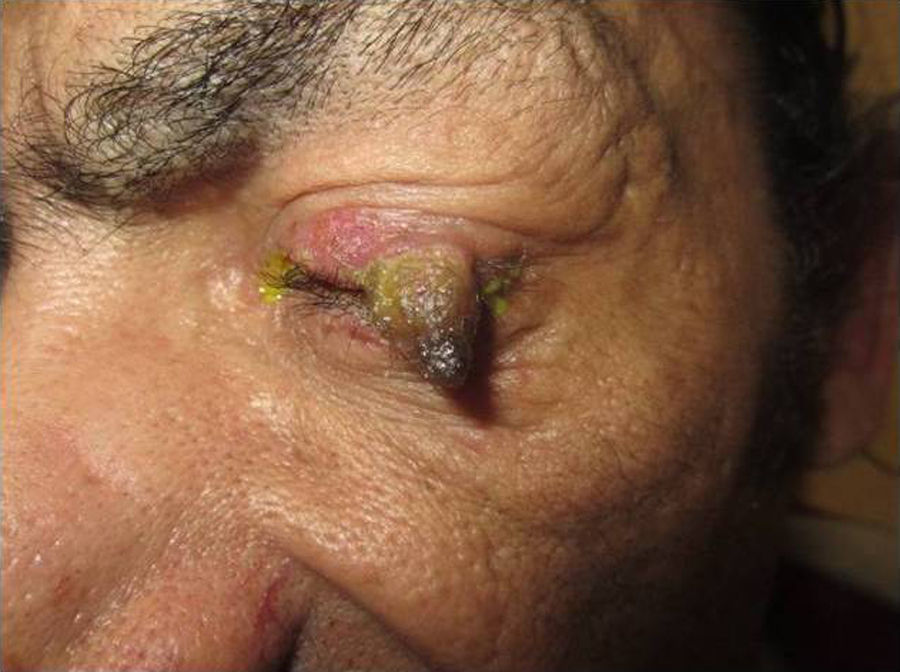
A panoramic image (Fig. 2) revealed a neoformation with a polypoid pattern that ulcerated the entire epidermis. This infiltrating tumor formed confluent nests that included some necrotic areas in the central zone. Higher magnification revealed the presence of cells with moderate atypia and prominent nucleoli in the tumor nests. Clear cell differentiation with a clear and finely vacuolated cytoplasm was also observed, and staining was positive for carcinoembryonic antigen, epithelial membrane antigen, and adipophilin.
Additional Tests
A full ophthalmologic examination revealed no pathological findings. Blood tests and chest radiograph were normal.
What Is Your Diagnosis?
DiagnosisSebaceous carcinoma.
Clinical Course and TreatmentThe lesion was removed by wedge excision with a surgical margin of 4mm. Margins were tumor-free in the histologic study. A computed tomography scan of the head, neck, and chest revealed no evidence of locoregional or distant metastases. No signs of tumor recurrence have been found during periodic follow-up over the past 2 years.
CommentSebaceous carcinoma (SC) is a rare, aggressive neoplasm of adnexal origin. SC accounts for 0.7% of all skin neoplasms.1 The most common site is the upper eyelid (75% of cases reported), although it can be found on any body part where sebaceous glands are present.2 SC usually presents as painful yellowish-pink nodules that may clinically resemble chalazions.3 In rare cases, it presents as a pedunculated mass resembling a cutaneous horn, as in our patient. Histology can show changes ranging from a well-differentiated sebaceous neoplasm with a lobular proliferation of neoplastic cells with abundant foamy cytoplasm—similar to mature sebocytes—to an undifferentiated tumor with an infiltrative growth pattern formed by neoplastic cells with marked nuclear pleomorphism, a high mitotic index, and a small amount of intracytoplasmic lipids. Stains such as adipophilin and perilipin have shown high sensitivity and specificity for this type of neoplasm.4 Metastases most frequently affect the local lymph nodes (preauricular, parotid, submandibular, and cervical). SC can spread through distant metastasis to organs such as the lungs, liver, bone, and brain.
SC can appear spontaneously or in the context of Muir-Torre syndrome. This syndrome has an autosomal dominant inheritance pattern and is associated with 1 or more sebaceous skin tumors (benign or malignant, as in the case of SC), the possible presence of keratoacanthomas, and 1 or more visceral malignancies, most frequently hereditary nonpolyposis colorectal cancer, followed by genitourinary tumors. Twenty-three percent of patients with Muir-Torre syndrome have SC, so it is important to rule out this association.
Surgery is the first-line treatment for SC. Because the eyelid is the most common site, the first-line technique is Mohs surgery,5,6 which is associated with a lower tumor recurrence rate (11%) than that of conventional surgery with 5 to 6mm safety margins (30%). Radiotherapy has been shown to be a good alternative to surgery.
The prognosis is very unfavorable, with a mortality rate of 50% in the first 5 years. Factors associated with poor prognosis include female sex, advanced age, time since onset of more than 6 months, tumor size >1cm and, histologically, an infiltrative tumor, poor differentiation, vascular or perineural invasion, and pagetoid dissemination.
Please cite this article as: Navarro-Triviño FJ. Tumor queratósico palpebral de largo tiempo de evolución. Actas Dermosifiliogr. 2017;108:667–668.


