



INTRODUCCION
El síndrome POEMS se caracteriza por la existencia de polineuropatía (P), organomegalia (O), endocrinopatía (E), componente M monoclonal (M) y alteraciones cutáneas (S, del inglés skin)1-3. Las manifestaciones cutáneas más frecuentes del síndrome son hiperpigmentación cutánea, edema, hipertricosis y rasgos esclerodermiformes, que afectan fundamentalmente a las zonas acras del cuerpo. Se han descrito también telangiectasias, acrocianosis, fenómeno de Raynaud, xerosis, hiperhidrosis, leuconiquia y lipodistrofia. Sólo los angiomas tuberosos pueden considerarse característicos del síndrome. Suelen ser múltiples, de aparición rápida, y localizados preferentemente en el tronco y la raíz de las extremidades.
DESCRIPCION DEL CASO
Una mujer de 54 años de edad consultó por presentar lesiones asintomáticas de aparición brusca en el tórax y en la cara, desde hacía 3 meses. Se trataba de pápulas eritematovioláceas, de 0,5 y 1 cm de tamaño y de consistencia blanda (figs. 1 y 2). El estudio anatomopatológico de una de las lesiones mostró dilataciones saculares de la dermis formando estructuras arracimadas (fig. 3).
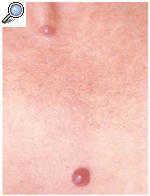
Fig. 1.--Nódulos eritematovioláceos en el tórax.

Fig. 2.--Nódulos eritematovioláceos en la cara.

Fig. 3.--Aspecto histológico de uno de los angiomas tuberosos. Aparecen espacios vasculares dilatados en la dermis, con acumulaciones de pequeños vasos sanguíneos, formando estructuras arracimadas, que se asemejan a los vasos sanguíneos glomerulares. (Hematoxilina-eosina, x100.)
La paciente había sido diagnosticada de síndrome POEMS en el año 1994 en el servicio de medicina interna porque presentaba un cuadro de síndrome constitucional, polineuropatía sensitivomotora grave de predominio distal, hepatosplenomegalia, poliadenopatías, hipotiroidismo subclínico, gammapatía monoclonal IgA-λ, acrocianosis y fenómeno de Raynaud. La paciente fue tratada inicialmente con varios ciclos de corticoides sistémicos y plasmaféresis, observándose una reducción de los niveles de interleucina 6 (IL-6) y mejoría de la sintomatología, especialmente de la polineuropatía. La paciente estaba realizando desde hacía 3 años tratamiento sistémico con labetalol, tiroxina, prednisona a dosis de 5 mg/día, carbonato cálcico, furosemida, bicarbonato sódico, alopurinol, eritropoyetina subcutánea, hierro y cianocobalamina en dosis de 1 mg intramuscular cada semana.
DISCUSION
El síndrome POEMS, también denominado síndrome de Crow-Fukase, síndrome de Shimpo, síndrome de Takatsuki y síndrome PEP (polineuropatía [P], endocrinopatía [E] y alteración de células plasmáticas [P]) presenta un amplio rango de manifestaciones y puede afectar a cualquier órgano.
Su etiopatogenia es desconocida. El desequilibrio entre las citocinas proinflamatorias (IL-6, IL-1β, y factor de necrosis tumoral alfa [TNF-α]) y las citocinas antiinflamatorias (factor del crecimiento intraepidérmico beta [TGF-β1]) podría explicar las características clínicas del síndrome. El hecho de que tras el tratamiento del mieloma se produzca una mejoría espectacular de la sintomatología sugiere que existen factores solubles responsables de los aspectos extramedulares del síndrome4. Dichos factores podrían actuar sobre la pared de los vasos sanguíneos provocando un aumento de la permeabilidad vascular. Podría tratarse de citocinas proinflamatorias, entre las que se implica especialmente a la IL-6 como principal reflejo de la actividad de la enfermedad5,6.
El objetivo del tratamiento es mejorar la polineuropatía. Para ello se ha utilizado la cirugía, la radioterapia, la quimioterapia, los corticoides sistémicos, las inmunoglobulinas intravenosas, la plasmaféresis y el trasplante autólogo de médula ósea7. En el caso de lesiones óseas únicas o escasas, se pueden emplear la cirugía y la radioterapia y en lesiones óseas difusas, se deben realizar tratamientos con quimioterápicos (melfalán o ciclofosfamida) o con corticoides sistémicos.


