
INTRODUCCIÓN
El síndrome de Muir-Torre (SMT) es una rara entidad, caracterizada por la aparición de tumores cutáneos derivados de las glándulas sebáceas, asociados a neoplasias malignas internas. Fue inicialmente descrito por Muir en 19671 y posteriormente por Torre en 19682. El número de casos descritos en la literatura hasta la actualidad supera escasamente los 200, pero probablemente sea un síndrome infradiagnosticado y su frecuencia sea mayor.
Actualmente se acepta como una genodermatosis de transmisión autosómica dominante, con penetrancia y expresividad variables3, 4. Recientemente ha sido descrita en asociación a otras enfermedades de base genética, como déficit de alfa-1-antitripsina5 e hiperlipidemia familiar6.
La detección de una historia familiar de carcinoma en estos pacientes4, 7, 8 ha llevado a encuadrar al SMT dentro del síndrome de carcinomatosis múltiple familiar (SCF) o síndrome de Linch II, como una manifestación fenotípica peculiar de éste, recomendándose la realización de despistaje en familiares asintomáticos, dado el riesgo de desarrollo de procesos neoplásicos en diferentes órganos9-12.
Inicialmente existió gran controversia sobre qué tipos de proliferaciones sebáceas debían ser incluidas en el síndrome hasta que Cohen et al13, 14 establecieron los criterios diagnósticos, aceptando como únicos marcadores cutáneos el adenoma, el epitelioma sebáceo o sebaceoma y el carcinoma sebáceo.
CASO CLÍNICO
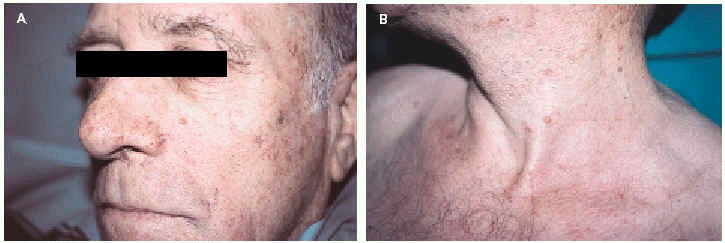
Un varón de 65 años con antecedentes de adenocarcinoma de colon, tratado quirúrgicamente en dos ocasiones (años 1981 y 1992), y adenocarcinoma gástrico, intervenido en 1995, fue remitido tras el diagnóstico de su primera neoplasia visceral a nuestra consulta por presentar varias lesiones cutáneas en el área facial. Entre los años 1992 y 1998 desarrolló 6 lesiones papulosas y papulonodulares, de coloración variable, amarillentas a marronáceas, de tamaño variable entre 0,3 y 0,8 cm de diámetro, localizadas en frente, mejilla izquierda, nariz y cuello (figs. 1A y B). Tras la exéresis, el diagnóstico histológico fue de dos sebaceomas, un adenoma sebáceo y tres hiperplasias sebáceas (figs. 2 y 3). Con la asociación de neoplasias viscerales y tumores de estirpe sebácea se estableció el diagnóstico de síndrome de Muir-Torre (SMT). El paciente evolucionó favorablemente, sin signos de recidiva ni metastásis.
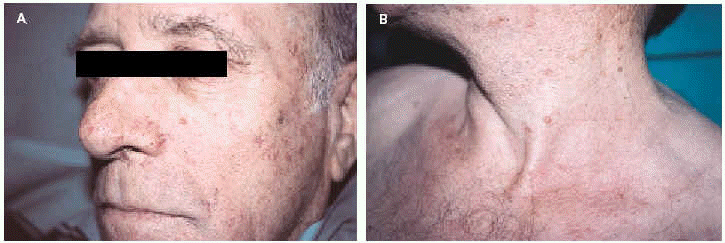
Fig. 1.--A: sebaceoma en mejilla izquierda. B: lesión papulosa amarillenta en cuello correspondiente a un adenoma sebáceo.
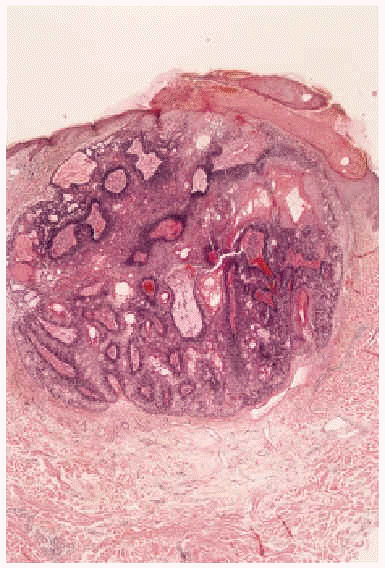
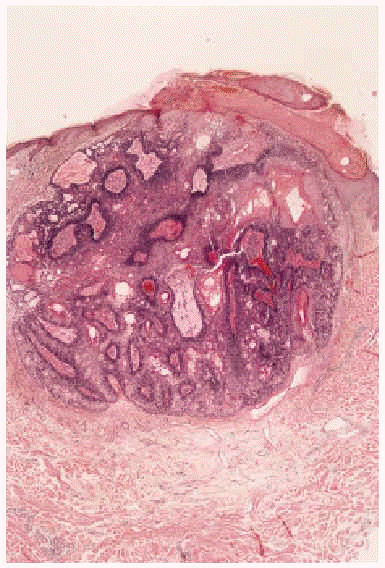
Fig. 2.--Imagen histológica del sebaceoma: nódulo polilobulado, bien delimitado en dermis superficial, con presencia de células basaloides inmaduras y nidos centrales de células sebáceas maduras sin atipia citológica (hematoxilina-eosina, 40x).
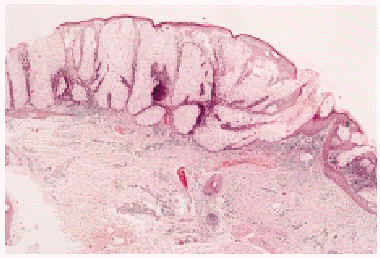
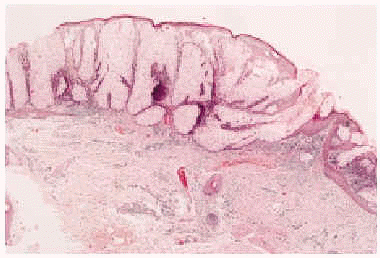
Fig. 3.--Imagen histológica del sebaceoma, apreciándose lóbulos sebáceos, de tamaño irregular, con predominio de células sebáceas maduras incompletamente diferenciadas, rodeadas de una capa de células basaloides (hematoxilina-eosina, 40x).
Existe además una importante historia de cáncer familiar por vía paterna, destacando padre, un tío, un primo y un sobrino fallecidos por neoplasias no filiadas, dos primos fallecidos por carcinomas de colon y estómago y un primo en tratamiento por adenocarcinoma de colon.
DISCUSIÓN
El adenoma sebáceo es el tumor más frecuente en el SMT3, 7, 15; se localiza fundamentalmente en el área facial, no metastatiza y no recidiva. Aunque semejantes datos clínicos aparecen también descritos en la población normal, hay que destacar el desarrollo de múltiples adenomas en otras localizaciones, no faciales, en los pacientes afectados de SMT15, 16. El epitelioma sebáceo tiene un comportamiento benigno, y aunque puede invadir localmente, el riesgo de metástasis es mínimo15, 17, 18. El carcinoma sebáceo, originado a partir de las glándulas de Meibomio, suele localizarse a nivel oculopalpebral, siendo importante destacar su menor poder metastatizante frente a localizaciones extraoculares1, 19-22. Las hiperplasias sebáceas se asocian frecuentemente, aunque no se consideran un marcador diagnóstico. Suelen aparecer en personas de edad avanzada, pero no implican un aumento de la incidencia de neoplasias viscerales malignas15, 16, 23. Los queratoacantomas aparecen descritos en un 20% de los pacientes con SMT, mostrando predilección por la edad media y localizándose en áreas fotoexpuestas3.
En este amplio espectro de proliferaciones sebáceas presentes en el SMT se incluyen, además, el hamartoma quístico foliculosebáceo24, 25, el epitelioma basocelular con diferenciación sebácea26, el tricofoliculoma sebáceo27 y el sebomatricoma, nueva denominación establecida por Sánchez Yus et al para un amplio grupo de lesiones cutáneas benignas con diferenciación sebácea28. No obstante, ninguna de las clasificaciones existentes es enteramente satisfactoria, agrupándose algunas bajo la denominación genérica de proliferación o lesión sebácea29. Otro hecho que dificulta el correcto diagnóstico es la ausencia de correlación clinicohistológica que frecuentemente se observa9.
Algunos autores propusieron como origen de determinados tumores derivados del complejo pilosebáceo, un fenómeno paraneoplásico inducido por algunas proteínas específicas producidas por tumores viscerales30. En la actualidad, el análisis genético molecular está esclareciendo algunos aspectos etiopatogénicos del SMT. Se acepta como una variante del SCF o del «cáncer colorrectal familiar no polipósico» (CCFNP), pues un subgrupo es alélico con ambas entidades10, 31, 32. Tanto en el SMT como en el CCFNP existe un error en la replicación genómica conocido como inestabilidad microsatélite, originada por mutaciones en la línea germinal heterozigota en los genes encargados de la reparación del ADN. Si además aparecen mutaciones somáticas adicionales en otros alelos, se pierde la capacidad para la reparación y aparece una clara predisposición al desarrollo de tumores en diferentes tejidos31, 33-35. Si los oncogenes y/o genes supresores también mutan, se desarrolla la neoplasia31, 36.
En el SMT las mutaciones se localizan fundamentalmente en la línea germinal h MSH2, y en algún caso aislado en h MLH1, mientras que en el CCFNP el trastorno genético afecta a ambos genes de forma similar y con menor frecuencia a las líneas germinales h PMS1 y h PMS237-41. La razón de la afectación predominante del epitelio colónico y de las glándulas sebáceas es un hecho aún no esclarecido31.
En una reciente revisión de la literatura se han descrito 205 casos de SMT con 399 neoplasias viscerales42. El número de tumores viscerales diagnosticados en cada paciente es variable y oscilan entre 1 y 9, aunque suelen ser únicos20. La relación temporal entre la aparición de las lesiones cutáneas y el diágnostico del cuadro neoplásico visceral es variable. El diagnóstico de la neoplasia interna fue previa a la del tumor sebáceo en el 56%, posterior en el 22% y simultáneo en el 6%. En el 16% de los casos publicados no se estableció una relación temporal42. El dermatólogo, por tanto, tiene un papel fundamental en el diagnóstico precoz del MTS ya que la detección de un adenoma, un epitelioma o un carcinoma sebáceo debe alertar sobre la posibilidad del desarrollo, ya establecido o posterior, de neoplasias viscerales. Son características asociadas al SMT una evolución lenta del proceso neoplásico sistémico, una capacidad metastatizante menor que en pacientes oncológicos no afectos de esta entidad y una larga supervivencia, con una media de 12 años a partir del diagnóstico3.
El carcinoma colorrectal es el tumor asociado con mayor frecuencia (aproximadamente el 60% del total)42. Presenta algunas particularidades, tales como su inicio en edades tempranas y su localización proximal al ángulo esplénico, a diferencia de la población normal, en la que el colon distal y el recto son los tramos más comúnmente afectos15, 42. El 28% de los pacientes desarrollan, además, poliposis colónica, asociación que constituye un marcador de riesgo para la aparición de otras neoplasias digestivas43.
En el 20%-25% de los casos aparecen tumores genitourinarios en todas sus localizaciones42. Los órganos implicados con mayor frecuencia son útero, riñón, ovario y próstata3. Menos habitual es la presencia de carcinoma de mama y procesos hematológicos, como linfomas Hodgkin y no Hodgkin, leucemia linfática crónica y policitemia vera3, 42, 44. Excepcionalmente, el SMT se desarrolla en el contexto de tumoraciones de cabeza, cuello, intestino delgado, estómago y páncreas, o asociada a melanoma3, 45.
La existencia de múltiples familiares afectos de neoplasias internas en un paciente afecto de SMT es frecuente, considerándose éste una expresión fenotípica más completa del SCF. Recientemente hemos diagnosticado en nuestro hospital otro SMT asociado a linfoma gástrico no Hodgkin primario sin infiltración linfomatosa en otros órganos, hecho clinicopatológico de gran interés, pues es una asociación previamente no descrita, coexistiendo además una significativa incidencia tumoral familiar46.
Es importante remarcar la conveniencia de la realización de un estudio sistémico completo y un seguimiento posterior a todo paciente diagnosticado de una o varias tumoraciones cutáneas de origen sebáceo dada su posible asociación a neoplasias viscerales internas. Al tratarse de un cuadro genético debe estudiarse la mayor cantidad posible de familiares de pacientes afectos, incluso asintomáticos, ya que en ellos el riesgo de desarrollar neoplasias internas malignas es mayor que en el resto de la población.


