


INTRODUCCION
El síndrome cardiofaciocutáneo es una entidad descrita inicialmente por Reynolds et al1 en 1986. Se caracteriza por presentar retraso psicomotor y del crecimiento, defectos cardiacos congénitos (principalmente estenosis pulmonar y defectos del tabique auricular), cara peculiar y múltiples anomalías ectodérmicas1-3. Se ha señalado que el síndrome cardiofaciocutáneo presenta una gran similitud con el síndrome de Noonan y el síndrome de Costello, y existe cierta controversia en la literatura médica sobre si se trata de entidades diferentes o variantes de un mismo síndrome3.
A continuación se describe un nuevo caso de síndrome cardiofaciocutáneo en el que destacan las manifestaciones cutáneas que acompañan al síndrome, y en particular su asociación con acantosis nigricans en axilas.
DESCRIPCION DEL CASO
Paciente varón de 17 años, diagnosticado de síndrome cardiofaciocutáneo, que consultó en el servicio de dermatología para la valoración de lesiones cutáneas. Sus padres estaban sanos y no eran consanguíneos; una hermana estaba sana. El paciente nació de un parto a término tras gestación normal, con peso de 3.450 g y Apgar 9/10. El cariotipo y cribado de metabolopatías fue normal. En el momento de la consulta presentaba como rasgos generales y hallazgos más significativos las siguientes manifestaciones: retraso mental moderado, talla de 160,7 cm (la talla genética que le correspondería sería de 174 cm), miocardiopatía hipertrófica (intervenido 10 años antes de estenosis subaórtica grave y subpulmonar con hipertrofia infundibular a expensas del conducto interventricular), anomalías en el fenotipo craneofacial (macrocefalia, frente alta, constricción bitemporal, depresión de raíz nasal y lóbulos auriculares grandes y rotados hacia atrás), ligera exoftalmia y nistagmo, miopía intensa (9 dioptrías), cataratas, pterigium colli bilateral, hernia inguinal derecha y criptorquidia del mismo lado, cifoescoliosis, cubitus valgus con imposibilidad de extensión completa de los codos, y manos con dedos gruesos, algo cortos e hipermóviles, al igual que los de los pies, que además mostraban hallux valgus.
Desde el punto de vista cutáneo presentaba numerosas pápulas foliculares eritematosas, algunas de ellas con hiperqueratosis central, de pequeño tamaño (1-3 mm), distribuidas principalmente en cejas, mejillas y caras de extensión de extremidades; estas lesiones se habían desarrollado en la infancia y en su evolución ocasionaron alopecia en cejas (fig. 1). Se apreciaba también de forma difusa en cara, tronco y raíz de miembros, diversas lesiones pigmentarias, de coloración marrón oscura, algo sobreelevadas, cuyo tamaño oscilaba entre 0,5 a 1 cm de diámetro y que correspondían histológicamente a nevos melanocíticos compuestos. En axilas la piel aparecía hiperpigmentada, de color marrón claro, ligeramente engrosada y verrucosa, con textura aterciopelada tipo acantosis nigricans (fig. 2). Además, el paciente mostraba un cabello de aspecto ensortijado y de implantación occipital baja, con ausencia de cejas, escasez de pestañas y falta de vello axilar.
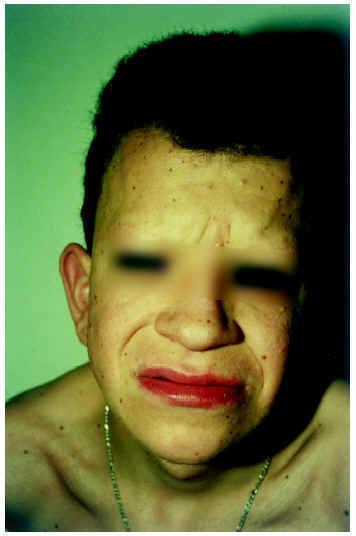
Fig. 1.--Anomalías en el fenotipo craneofacial y alopecia en cejas.
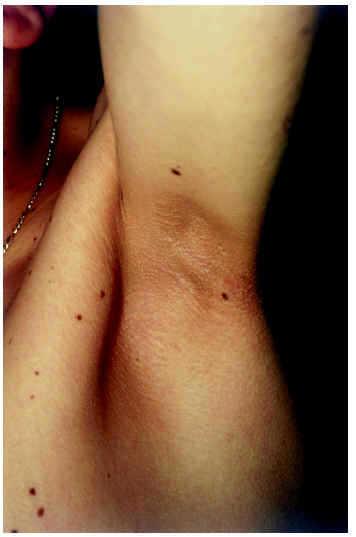
Fig. 2.--Piel hiperpigmentada, ligeramente engrosada y verrucosa, tipo acantosis nigricans en axilas.
DISCUSION
Las lesiones cutáneas en el síndrome cardiofaciocutáneo son muy variables y se han descrito hasta en el 95 % de los casos4. La presentación más característica consiste en lesiones de tipo queratosis pilar o pápulas foliculares, rodeadas de halo eritematoso, que se localizan principalmente en cara y superficie extensora de miembros; cuando afectan a las cejas y provocan alopecia por destrucción folicular se conoce con el nombre de uleritema ofriógenes. Esta entidad no es específica del síndrome cardiofaciocutáneo ya que también se ha considerado un marcador del síndrome de Noonan5,6, e incluso puede aparecer de manera aislada con transmisión probablemente autosómica dominante7. Otras manifestaciones cutáneas descritas en este síndrome son piel seca con aspecto ictiosiforme, eccemas, dermatitis seborreica, manchas «café con leche», nevos pigmentarios, angiomas, linfedema y piel redundante en manos y pies1-3,4,8.
Las alteraciones en el pelo son también comunes y se considera que aparecen en el 84-100 % de los casos4. Consisten en grados variables de alopecia con pelo de aspecto lanoso, rizado, frágil o quebradizo2,3,4,9. La línea de implantación posterior del cabello es baja y el pelo de las cejas y las pestañas está ausente o es escaso en más del 50 % de los casos4.
Mathews et al10 publicaron un caso que, junto a las manifestaciones cutáneas clásicas del síndrome, mostró como hallazgos más significativos una acantosis nigricans hipopigmentada en cuello y axilas y un siringocistoadenoma papilífero en muslo. El caso que nosotros describimos presenta como observación más peculiar las lesiones típicas de acantosis nigricans en axilas. En la revisión realizada por Wieczorek et al3, estos autores hallaron lesiones de acantosis nigricans en 2 de 44 casos (4,5 %), lo que coloca a esta anomalía cutánea en una presentación muy poco frecuente.
En conclusión se describe un nuevo caso de síndrome cardiofaciocutáneo en el que junto a las lesiones cutáneas ya clásicas de pápulas foliculares y ulecitema ofriógenes, nevos pigmentarios y alteraciones en el pelo, presentaba como hallazgo más destacable acantosis nigricans en axilas.


