



INTRODUCCION
Buschke y Ollendorff1 describieron por primera vez la asociación osteopoiquilia y lesiones cutáneas que denominaron dermatofibrosis lenticularis disseminata en 1928. Este es un síndrome raro, de herencia autosómica dominante, caracterizado por áreas hiperdensas en huesos asociado a nevos del tejido conjuntivo manifestados por lesiones infiltradas, amarillentas o del color de la piel normal que pueden coalescer y suelen afectar a muslos y glúteos y más raramente a las extremidades superiores, espalda y abdomen. Histológicamente existen varias formas: la más frecuente presenta aumento de fibras elásticas, pero en otros casos pueden estar conservadas, disminuidas o fragmentadas. También se observan anomalías en el colágeno.
Se presentan los casos de una mujer y su hija con nevos del tejido conjuntivo y osteopoiquilia asociada en la madre.
DESCRIPCION DEL CASO
Una mujer de 41 años presentaba, como antecedentes de interés, un síndrome de la silla turca vacía, cataratas y osteopoiquilia. Como antecedente familiar de interés su madre estaba diagnosticada de esclerodermia. La paciente presentaba lesiones cutáneas asintomáticas desde el nacimiento, de distribución simétrica y bilateral, en muñecas y tobillos, que consistían en placas del color de la piel de consistencia firme y aspecto rugoso (fig. 1). La biopsia cutánea de una lesión mostraba un leve engrosamiento de la dermis. Con la técnica de Van Gieson se observó aumento de las fibras elásticas de la dermis reticular, con haces gruesos ramificados, compatible con un nevo elástico (fig. 2).
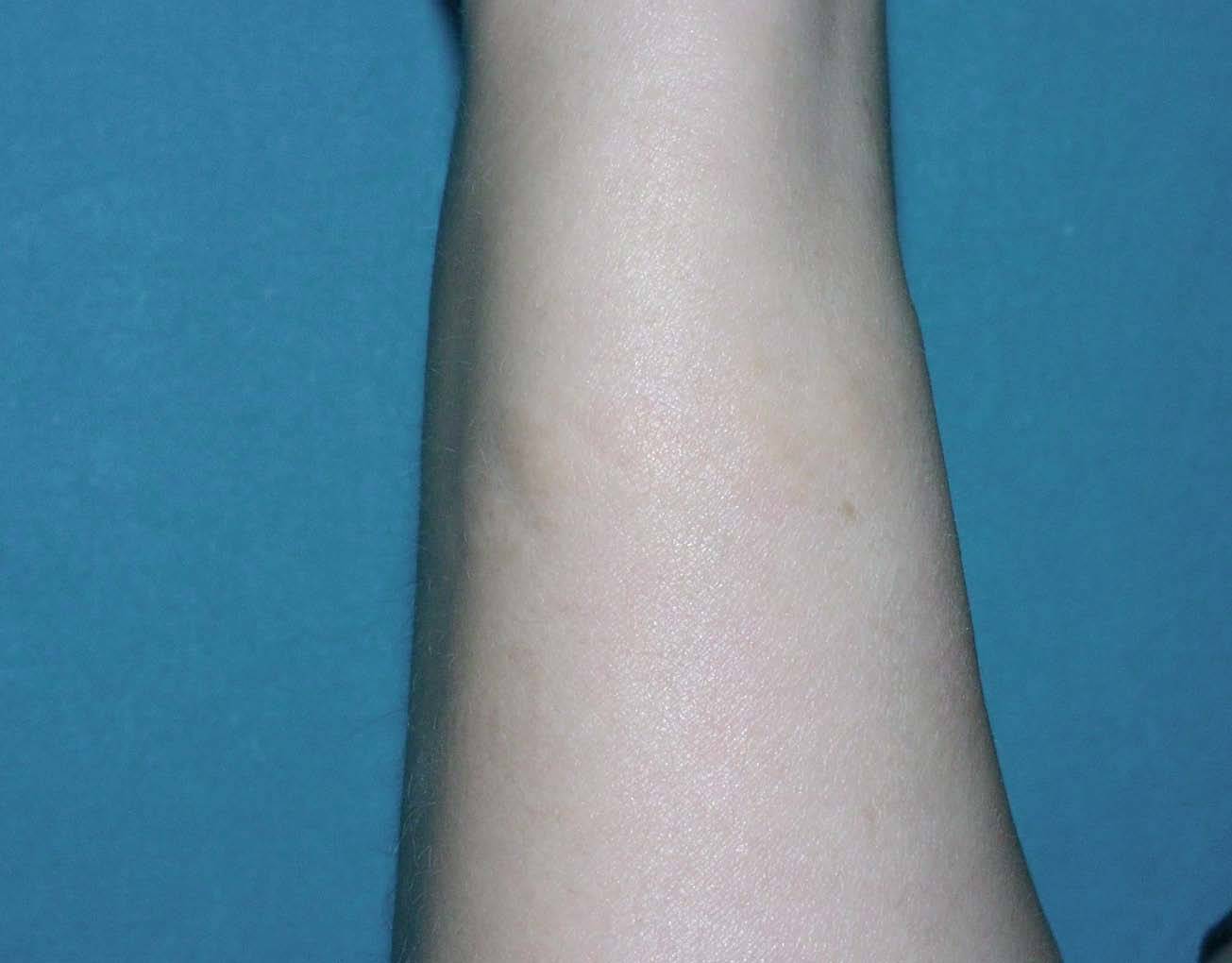
Fig. 1.--Antebrazo donde se observan pápulas del color de la piel.
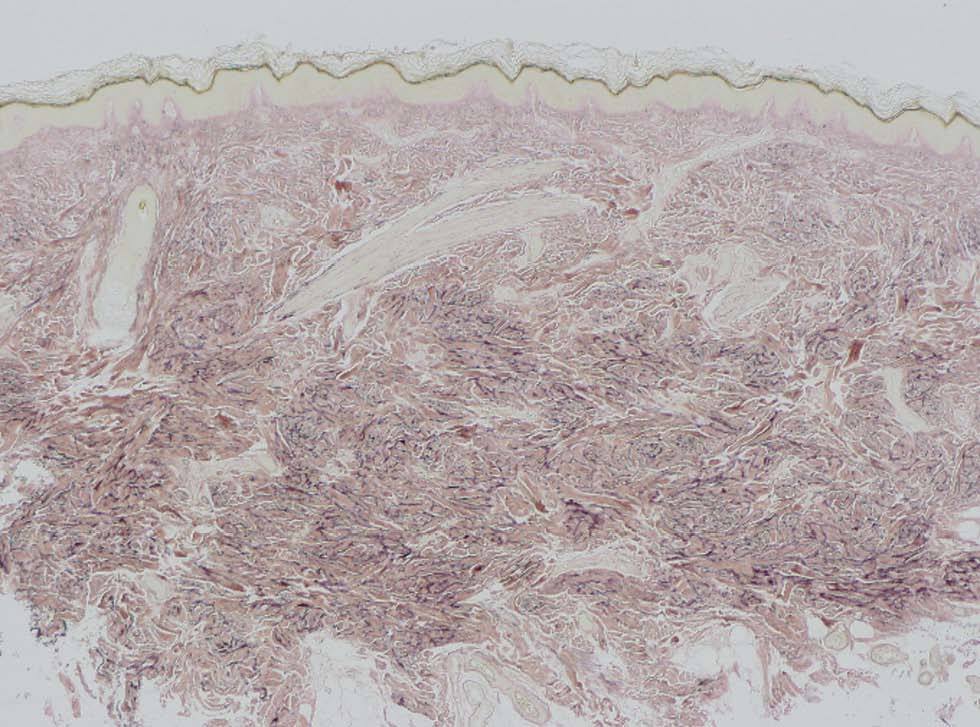
Fig. 2.--Aumento de fibras elásticas en la dermis reticular. (Van Gieson x4.)
La hija de la paciente de 2 años de edad presentaba lesiones similares a las de su madre, que comenzaron en los primeros meses de vida, asintomáticas en miembros inferiores, consistentes en pápulas amarillentas, de consistencia firme y rugosas (fig. 3).
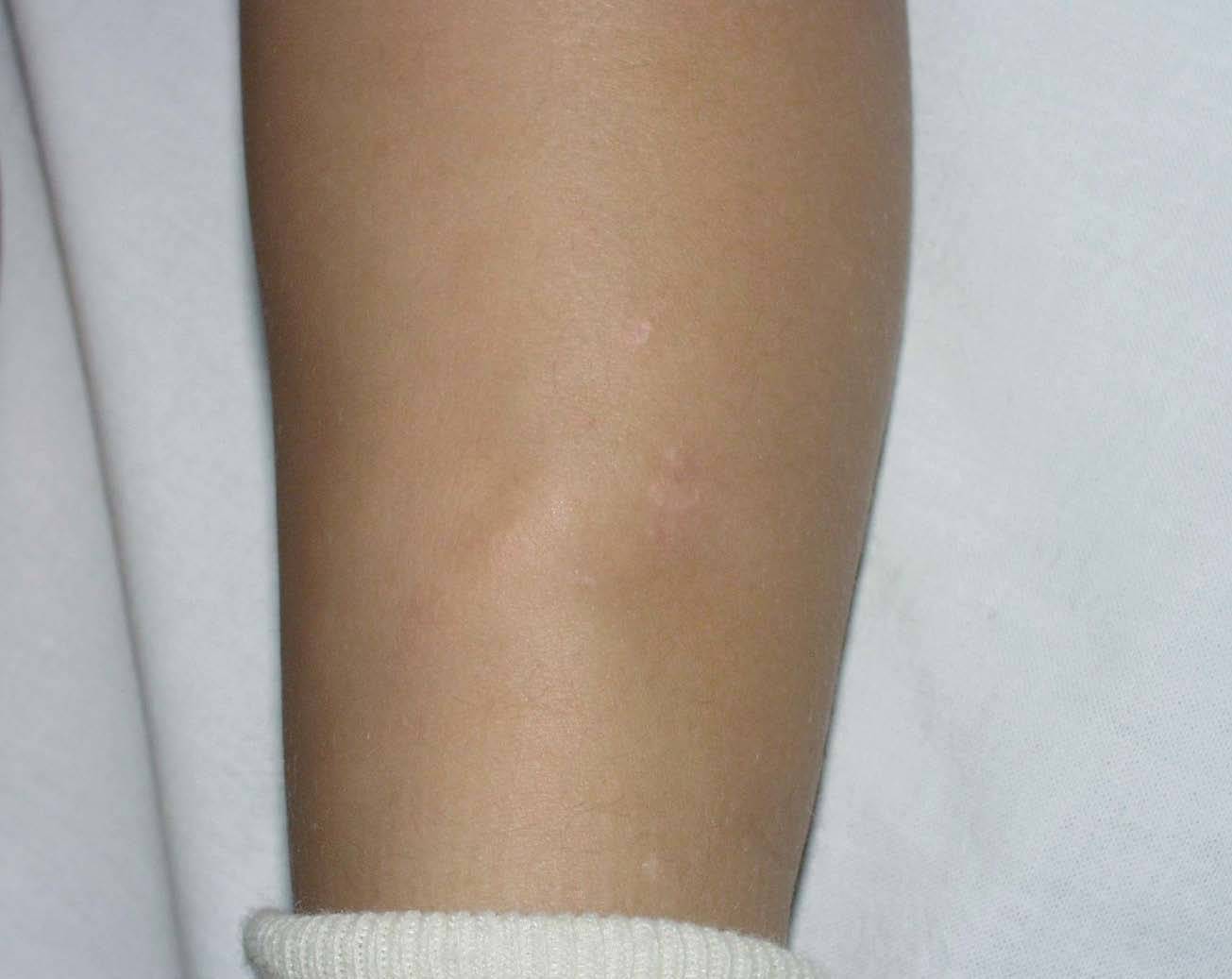
Fig. 3.--Pápulas agrupadas en la cara externa de la pierna.
La biopsia cutánea reveló una epidermis sin alteraciones, y en la dermis haces de colágeno de morfología y agrupación normales. La coloración para fibras elásticas demostró una gran pobreza de éstas en todos los niveles, compatible con nevo anelástico. No se le practicó estudio óseo radiológico.
DISCUSION
En el síndrome de Buschke-Ollendorff se asocian lesiones cutáneas denominadas dermatofibrosis lenticular diseminada, y lesiones óseas consistentes en áreas de densidad aumentada en la esponjosa de huesos del carpo, tarso y falanges, así como metáfisis y epífisis de huesos largos y en pelvis. Estas lesiones óseas son un hallazgo radiológico. Otras asociaciones descritas en este síndrome son úlcera péptica, cataratas, estrías, queloides, lentigos, fibromas orales, queratodermia palmoplantar estriada, anetodermia, retraso mental, acondrodisplasia, enfermedad de Dupuytren, síndrome de La Peyronie, morfea, esclerodermia y otras2-4. El defecto básico del síndrome parece estar causado por heterozigocidad para mutaciones que inducen pérdida de función en el gen LEMD3, también llamada MAN-1, que codifica una proteína de la membrana nuclear interna4.
En la misma familia puede haber miembros con osteopoiquilia sin nevos y viceversa5. Se ha descrito un síndrome de Buschke-Ollendorff abortado6,7 que se caracteriza por ausencia de malformaciones óseas y disminución del tejido elástico. Así, podemos considerar que nuestros pacientes constituyen un caso familiar de síndrome de Buschke-Ollendorff, si bien sería necesario hacer un rastreo óseo a la niña para completar el estudio. El antecedente de esclerodermia en la familia podría estar relacionado ya que es una de las asociaciones descritas en este síndrome4.


