


INTRODUCCIÓN
El síndrome de Bart fue descrito en una familia muy numerosa como una epidermólisis ampollosa de herencia autosómica dominante con penetrancia completa y expresividad variable (1). Sus características clínicas son ausencia congénita localizada de la piel (ACLP), con afectación fundamentalmente en las piernas, lesiones ampollosas mucocutáneas y alteraciones de las uñas, aunque no todos los pacientes presentan el cuadro clínico completo. Algunos autores consideraron que el síndrome de Bart no tenía una entidad propia, sino que correspondía a un subtipo de cualquiera de las formas de epidermólisis ampollosa que se asocie a una ausencia congénita localizada de piel (2). Tras el estudio de la familia original de Bart se demostró que los pacientes originales mostraban características ultraestructurales, inmunohistoquímicas y genéticas similares a las de la forma dominante de epidermólisis ampollosa distrófica (3, 4). Por tanto, el síndrome de Bart constituye un subtipo peculiar o variante alélica de la epidermólisis ampollosa distrófica dominante (EADD). Presentamos un caso clínico de ACLP en un niño con características clinicopatológicas de EADD con lesiones ungueales compatibles de su padre.
DESCRIPCION DEL CASO
Un niño de 6 días de edad, nacido tras parto a término, cefálico y espontáneo, presentaba desde el nacimiento una zona carente de piel en la región pretibial derecha y pérdida de la uña del primer dedo de la mano izquierda. Durante la primera semana de vida presentó ampollas dispersas en la piel en escaso número. No se referían lesiones en las mucosas. Entre los antecedentes familiares destacaba una onicodistrofia no filiada en el padre, sin antecedentes de lesiones ampollosas ni aplasia cutánea. A la exploración se observó un área de denudación completa, de forma alargada y bien delimitada, que ocupaba gran parte de la región pretibial y rodilla derechas (Fig. 1 A). Se apreció una ampolla de pequeño tamaño, hemorrágica, en la mano izquierda y varias costras en muslo izquierdo, cara y manos. No se apreciaron lesiones de mucosas y el resto de la exploración clínica fue normal. El padre del paciente presentaba en la mayor parte de las uñas de las manos y los pies una disminución de su tamaño asociada a una coloración amarillenta y grisácea de la lámina ungueal y engrosamiento de la misma.
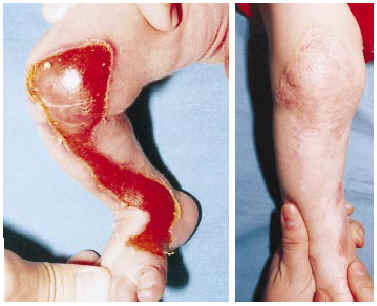
FIG. 1.--A: Ausencia congénita localizada de piel en región pretibial y rodilla derechas. B: Reepitelización completa de la lesión inicial con cicatrización distrófica y quistes miliares.
En el estudio histológico de una biopsia de una ampolla cutánea se observó un despegamiento dermo-epidérmico con escaso componente inflamatorio. La tinción con PAS reveló que la membrana basal quedaba adherida al techo de la ampolla. En el estudio ultraestructural se apreció que se producía un despegamiento por debajo de la membrana basal (Fig. 2). El niño fue tratado con mupirocina tópica y vendajes, con buena evolución clínica. A los 6 meses la aplasia cutánea había reepitelizado, dejando una cicatriz atrófica que se acompañaba de múltiples quistes miliares en la cicatriz y en el dorso de ambas manos (Fig. 1 B).

FIG. 2.--Microscopia electrónica que muestra un despegamiento ampolloso por debajo de la membrana basal.
DISCUSION
En 1966, Bart y cols. (1) describieron una familia que presentaba ausencia congénita localizada de la piel (ACLP), lesiones ampollosas en piel y/o mucosas y alteraciones de las uñas con un patrón de herencia autosómica dominante con penetrancia incompleta y variable expresividad. El artículo original no reseñaba ningún estudio histológico ni ultraestructural. Desde entonces, varios autores (2, 5-8) describieron casos de ACLP en pacientes con diversas formas de epidermólisis ampollosa (incluyendo la forma simple, distrófica recesiva, distrófica dominante y letal de Herlitz), considerando que el término de síndrome de Bart debería ser empleado para cualquier forma de epidermólisis ampollosa asociada con ausencia congénita localizada de piel. En estos casos se postuló que la aplasia cutánea era debida a un traumatismo intraútero. Posteriormente se revisó la familia original, realizándose un estudio ultraestructural, inmunohistoquímico y genético, que demostraron que dicha familia presentaba características de epidermólisis ampollosa distrófica dominante (3, 4). El estudio ultraestructural de la piel perilesional mostraba una disminución de las fibrillas de anclaje y el clivaje se localizaba por debajo de la lámina densa. En la inmunohistoquímica se emplearon anticuerpos contra el colágeno tipo IV que se localizaron en el techo de la ampolla, mientras que el colágeno tipo VII presentaba una distribución normal. En el estudio de ligamento genético se detectó que la enfermedad estaba ligada al gen del colágeno tipo VII (COL7A1) que se encuentra en el brazo corto del cromosoma 3 (3p21) y en el análisis de mutaciones se observó la sustitución de una glicina por una arginina en el exon 73 del gen del colágeno tipo VII (COL7A1). Por tanto se demostró que el denominado síndrome de Bart es una variante fenotípica de la epidermólisis ampollosa distrófica dominante.
Clínicamente presenta una expresividad variable y no todos los pacientes exhiben el cuadro clínico completo (1, 2, 9). La ausencia congénita localizada de la piel suele ser bilateral y con frecuencia simétrica, aunque existen casos unilaterales. La localización más frecuente son las piernas, en particular la región pretibial. Algunos autores postulan que la ACLP podría seguir las líneas de Blaschko (10). Las lesiones ampollosas se inician después del nacimiento, durante el período neonatal, en áreas de piel sometidas a presión o traumatismo y suelen disminuir a partir del primer año de vida. En ocasiones se observan cicatrices, hipopigmentación y quistes miliares, aunque en la mayoría de las ocasiones curan sin secuela. Es frecuente que la afectación de la mucosa oral coincida con el comienzo de la alimentación con la tetina de plástico y en raras ocasiones se extiende al esófago, faringe y/o tracto respiratorio superior. Aunque en estos casos pueden producirse regurgitación, pérdida del apetito o problemas respiratorios, éstos suelen resolverse sin secuelas. Las alteraciones de las uñas ocurren típicamente en los niños con cuadros más extensos. Los pies, especialmente la uña del primer dedo, se afectan con mayor frecuencia que las manos, en forma de ausencia congénita, distrofia o pérdida de la uña. En raras ocasiones se han asociado otras anomalías como aplasia renal, venas varicosas y trastornos congénitos en las extremidades inferiores.
El tratamiento es conservador, con un manejo cuidadoso del niño y prevención de infecciones cutáneas secundarias empleando antibióticos tópicos. El pronóstico es muy bueno, sin afectación del crecimiento y desarrollo del niño, con reepitelización de la aplasia cutánea a las pocas semanas. Los brotes de lesiones ampollosas suelen desaparecer después de la pubertad y las lesiones ungueales generalmente persisten de por vida (2, 9).


