
















Los sarcomas constituyen un grupo amplio de tumores, muchos de ellos con comportamiento biológico y agresividad diferentes entre sí, que habitualmente requieren un tratamiento multidisciplinario, frecuentemente complejo. El desarrollo en las últimas décadas de la dermatología quirúrgica y oncológica ha permitido que los dermatólogos se conviertan en los especialistas responsables del diagnóstico y tratamiento del cáncer cutáneo. El propósito de este artículo es revisar los principales sarcomas de partes blandas de localización típicamente cutánea.
El dermatofibrosarcoma protuberans es un sarcoma de bajo grado de malignidad, con un crecimiento lento e infiltrativo localmente y escasa capacidad metastásica (<3%). El tratamiento de elección es la cirugía micrográfica de Mohs. Es recomendable solicitar el estudio de la translocación COL1A1-PDGFB cuando existen dudas diagnósticas, y para determinar qué pacientes pueden responder a los fármacos inhibidores de la tirosina quinasa. El imatinib está indicado en el dermatofibrosarcoma protuberans localmente avanzado y metastásico.
Sarcomas comprise a broad group of tumors, many of whose biological behavior and aggressiveness differ from one type to another. The therapeutic approach is generally multidisciplinary and often complex. Developments in surgical and oncological dermatology during the last few decades have positioned dermatologists as specialists in the diagnosis and treatment of skin cancer. The aim of this article is to review the main soft tissue sarcomas that typically affect the skin. Dermatofibrosarcoma protuberans is a low-grade malignant sarcoma. It exhibits slow-growth, is locally invasive, and has low metastatic potential (<3%). Mohs micrographic surgery is the treatment of choice. The COL1A1-PDGFB translocation should be analyzed in cases of unclear diagnosis and when it is necessary to identify candidates for tyrosine kinase inhibitors. Imatinib is indicated for the treatment of locally advanced and metastatic dermatofibrosarcoma protuberans.

Los sarcomas constituyen un grupo amplio y heterogéneo de tumores poco comunes, que habitualmente requieren un tratamiento multidisciplinario, frecuentemente complejo. El desarrollo en las últimas décadas de la dermatología quirúrgica y oncológica ha permitido que los dermatólogos se conviertan en los especialistas responsables del diagnóstico y tratamiento del cáncer cutáneo. Este aspecto es especialmente importante en el caso de los sarcomas de partes blandas localizados en la piel. En la actualidad existen diversas guías de tratamiento de sarcomas, pero que no describen específicamente los sarcomas cutáneos. Además, estas directrices han sido realizadas por expertos en las diversas especialidades que se ven implicados en las diferentes etapas del diagnóstico y tratamiento de los sarcomas (oncólogos, patólogos, radiólogos, traumatólogos, cirujanos generales y plásticos), pero no se ha tenido en cuenta la participación de los dermatólogos. Este hecho es fundamental, ya que en muchas ocasiones el tratamiento de un sarcoma localizado en el retroperitoneo no es extrapolable a un sarcoma localizado en la piel. El objetivo de este artículo es proporcionar unas recomendaciones claras, basadas en la mayor evidencia clínica posible, del manejo de los principales sarcomas cutáneos desde el punto de vista dermatológico que faciliten una buena práctica clínica.
GeneralidadesLos sarcomas constituyen un amplio grupo de tumores, muchos de ellos con comportamiento biológico y agresividad diferentes entre sí. Los sarcomas de partes blandas son un grupo heterogéneo y poco frecuente de tumores malignos de origen mesenquimal, que suponen menos del 1% de todos los tumores malignos del adulto y el 12% de los cánceres de la edad pediátrica1,2. La mayor parte de los sarcomas (80%) se originan en los tejidos blandos (entre ellos la piel), mientras que el resto lo hacen en el hueso, y menos frecuentemente, en las vísceras.
El espectro histopatológico de los sarcomas de partes blandas es muy amplio, posiblemente porque las células embrionarias mesenquimales de las que se originan tienen capacidad de diferenciación hacia muchos otros tejidos. La clasificación de la Organización Mundial de la Salud de los sarcomas de partes blandas está basada en el posible origen tisular de las diferentes variedades de tumor, incluyendo entidades tales como el fibrosarcoma, el angiosarcoma, el liposarcoma, el leiomiosarcoma, el rabdomiosarcoma o el sarcoma sinovial. Esta clasificación contempla más de 100 subtipos histológicos1,2.
Los más frecuentes a nivel global son el sarcoma pleomórfico indiferenciado, seguido del liposarcoma, el leiomiosarcoma y el mixofibrosarcoma. A nivel cutáneo los más frecuentes son el dermatofibrosarcoma protuberans y el sarcoma de Kaposi3.
El diagnóstico y clasificación de los sarcomas de partes blandas se realiza en función de su patrón histológico, los hallazgos inmunohistoquímicos y las anomalías citogenéticas asociadas1,3,4. La histología continúa siendo el pilar fundamental del diagnóstico, mientras que la inmunohistoquímica permite orientar el origen histogenético del sarcoma. Actualmente también se lleva a cabo el diagnóstico molecular con técnicas de hibridación in situ con fluorescencia (FISH), la transcripción inversa y reacción en cadena de la polimerasa (RT-PCR) y la secuenciación, que permiten identificar translocaciones cromosómicas específicas de muchos sarcomas.
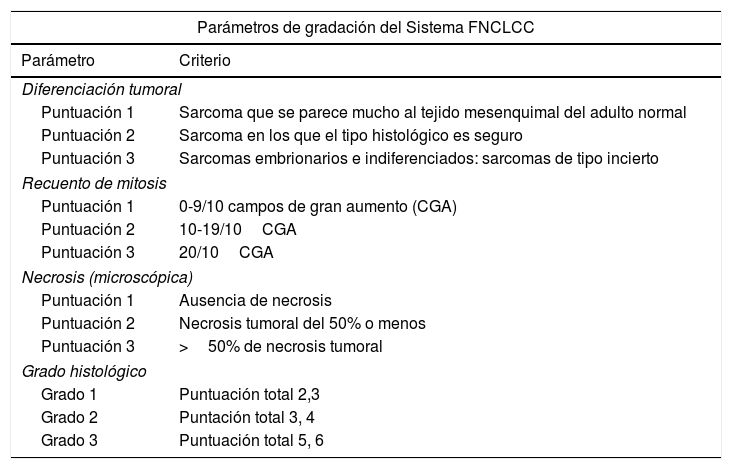
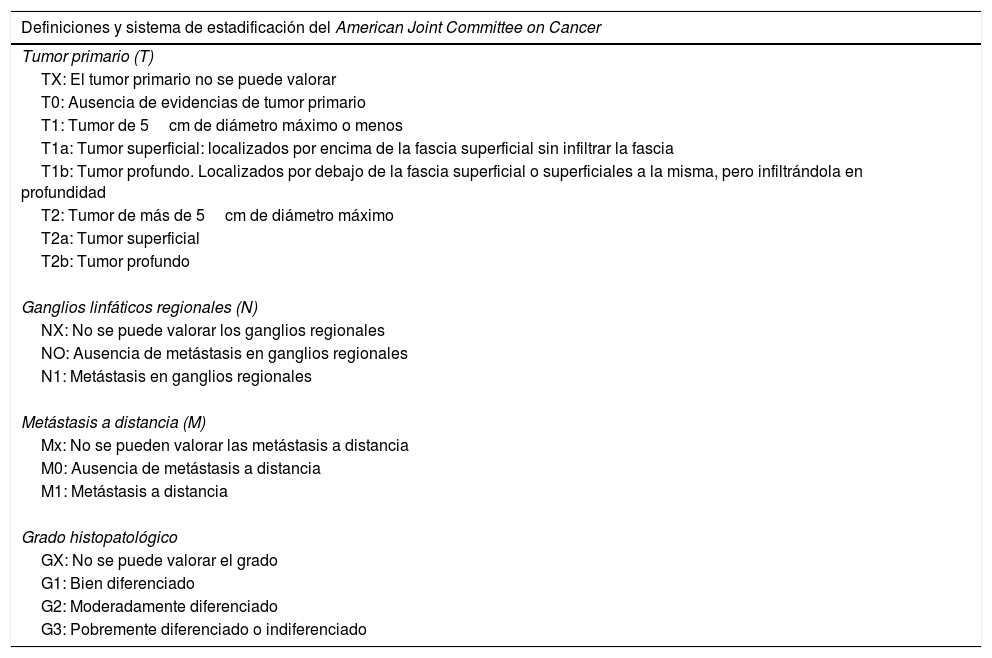
Se han desarrollado varios sistemas de gradación y de estadificación de los sarcomas, que tratan de predecir el pronóstico. Los 2 sistemas más utilizados en la actualidad son el de la Fédération National de Centres de Lutte Contre le Cancer (FNCLCC) de Francia (tabla 1) y el del American Joint Committe on Cancer (AJCC) (tabla 2)5–7. El sistema de gradación francés se basa en una valoración de los parámetros de diferenciación histológica, en el número de mitosis y en la presencia de necrosis, mientras que el de la AJCC está basado en el tamaño y localización del tumor (superficial o profundo), la afectación ganglionar, las metástasis y el grado de diferenciación histológico8,9.
Gradación de sarcomas de partes blandas del sistema de Fédération National de Centres de Lutte Contre le Cancer7
| Parámetros de gradación del Sistema FNCLCC | |
|---|---|
| Parámetro | Criterio |
| Diferenciación tumoral | |
| Puntuación 1 | Sarcoma que se parece mucho al tejido mesenquimal del adulto normal |
| Puntuación 2 | Sarcoma en los que el tipo histológico es seguro |
| Puntuación 3 | Sarcomas embrionarios e indiferenciados: sarcomas de tipo incierto |
| Recuento de mitosis | |
| Puntuación 1 | 0-9/10 campos de gran aumento (CGA) |
| Puntuación 2 | 10-19/10CGA |
| Puntuación 3 | 20/10CGA |
| Necrosis (microscópica) | |
| Puntuación 1 | Ausencia de necrosis |
| Puntuación 2 | Necrosis tumoral del 50% o menos |
| Puntuación 3 | >50% de necrosis tumoral |
| Grado histológico | |
| Grado 1 | Puntuación total 2,3 |
| Grado 2 | Puntación total 3, 4 |
| Grado 3 | Puntuación total 5, 6 |
Estadificación de los sarcomas de partes blandas (TNM) tomado del American Joint Committee on Cancer (AJCC) (7th ed, 2010) versión 20165
| Definiciones y sistema de estadificación del American Joint Committee on Cancer |
|---|
| Tumor primario (T) |
| TX: El tumor primario no se puede valorar |
| T0: Ausencia de evidencias de tumor primario |
| T1: Tumor de 5cm de diámetro máximo o menos |
| T1a: Tumor superficial: localizados por encima de la fascia superficial sin infiltrar la fascia |
| T1b: Tumor profundo. Localizados por debajo de la fascia superficial o superficiales a la misma, pero infiltrándola en profundidad |
| T2: Tumor de más de 5cm de diámetro máximo |
| T2a: Tumor superficial |
| T2b: Tumor profundo |
| Ganglios linfáticos regionales (N) |
| NX: No se puede valorar los ganglios regionales |
| NO: Ausencia de metástasis en ganglios regionales |
| N1: Metástasis en ganglios regionales |
| Metástasis a distancia (M) |
| Mx: No se pueden valorar las metástasis a distancia |
| M0: Ausencia de metástasis a distancia |
| M1: Metástasis a distancia |
| Grado histopatológico |
| GX: No se puede valorar el grado |
| G1: Bien diferenciado |
| G2: Moderadamente diferenciado |
| G3: Pobremente diferenciado o indiferenciado |
| Estadio | Grado | Tumor primario | Ganglios regionales | Metástasis |
|---|---|---|---|---|
| IA | G1 o GX | T1a o T1b | N0 | M0 |
| IB | G1 o GX | T2a o T2b | N0 | M0 |
| IIA | G2 o G3 | T1a o T1b | N0 | M0 |
| IIB | G2 | T2a o T2b | N0 | M0 |
| III | Cualquier G | Cualquier T | Cualquier N | M0 |
| IV | Cualquier G | Cualquier T | Cualquier N | M1 |
Los sarcomas de partes blandas tienden más a la diseminación hematógena que a la linfática. De hecho, la afectación ganglionar es poco frecuente, pero puede ser típica de algunas variedades como el rabdomiosarcoma, el sarcoma sinovial, el sarcoma de células claras o el sarcoma epitelioide. Los tumores de mal pronóstico por su tamaño, localización profunda o alto grado según FNCLCC originan metástasis generalmente pulmonares, que están presentes al diagnóstico en un 10% de los sarcomas de partes blandas6. No obstante, la mayor parte de los sarcomas de partes blandas de interés dermatológico tienen bajo riesgo de diseminación hematológica, a excepción del angiosarcoma y el leiomiosarcoma subcutáneo (tabla 3)6.
Principales sarcomas cutáneos clasificados en función del grado histológico junto con la supervivencia aproximada a los 5 años
| Tipo histológico | Grado Histológico | Pronóstico | ||
|---|---|---|---|---|
| I | II | III | Supervivencia 5 años | |
| Sarcoma de Kaposi | 60-100% depende del estado inmunitario | |||
| Dermatofibrosarcoma protuberans | 97-100% | |||
| Dermatofibrosarcoma-Fibrosarcomatoso | 90-95% | |||
| Fibrosarcoma congénito | 90-100% | |||
| Leiomiosarcoma | 97% dérmico 65% subcutáneo | |||
| Liposarcoma | 80% | |||
| Sarcoma pleomórfico | 80-90% | |||
| Sarcoma epitelioide | 70% | |||
| Tumor maligno de la vaina nerviosa periférica | 60% | |||
| Angiosarcoma | 35-40% | |||










El propósito de este artículo es revisar los sarcomas de partes blandas de localización típicamente cutánea, con especial énfasis en el dermatofibrosarcoma protuberans, el sarcoma dérmico pleomórfico, el leiomiosarcoma, el angiosarcoma y el sarcoma de Kaposi. Otros sarcomas con posible localización cutánea, como el sarcoma epitelioide, el tumor maligno de la vaina nerviosa y el liposarcoma se han omitido en esta revisión por su baja frecuencia en la consulta dermatológica.
El presente trabajo no pretende revisar exhaustivamente todos los sarcomas cutáneos y/o superficiales, sino ayudar a orientar al dermatólogo sobre el diagnóstico y tratamiento de las variantes más frecuentes.
Dermatofibrosarcoma protuberansIntroducciónEl dermatofibrosarcoma protuberans (DFSP) es un sarcoma de la piel, infrecuente, de crecimiento lento, generalmente indolente, que comprende un 5% de todos los sarcomas. Aproximadamente el 80-90% de ellos son de bajo grado y menos del 3% dan lugar a metástasis, pero las recaídas locales son frecuentes debido a su carácter infiltrativo. La extirpación quirúrgica completa es el tratamiento de elección. Los pacientes con DFSP presentan una supervivencia global a los 5 años muy alta (99-100%)10–12.
Epidemiología y diagnóstico clínicoEl DFSP representa menos del 0,1% de todos los tumores cutáneos10. Su incidencia se ha calculado entre 0,8 y 5 casos por millón de habitantes/año11. Aparece más frecuentemente en adultos jóvenes, entre la segunda y la quinta décadas de la vida, aunque puede aparecer a cualquier edad: desde el nacimiento hasta la vejez13,14. En cuanto a sexos parece existir una distribución igual entre hombres y mujeres. El DFSP aparece en todas las etnias, pero con una mayor frecuencia en la etnia negra, en especial la variedad pigmentada o tumor de Bednar. La localización más frecuente del DFSP es el tronco10, aproximadamente entre un 50% y un 60% se hallan en esta área. Otras zonas frecuentes son la parte proximal de las extremidades (20-30%) o la región cervicocefálica (10-15%), especialmente el cuero cabelludo, la frente o la fosa supraclavicular15,16.
El DFSP aparece como un tumor solitario, de superficie multilobulada y de forma y tamaño variables. La lesión muestra una consistencia muy firme a la palpación, adherido a la piel suprayacente, pero móvil sobre tejidos subyacentes17. El aspecto clínico del DFSP depende del tiempo de evolución. Se trata de un tumor de crecimiento muy lento, habiéndose descrito casos de más de 50 años hasta su diagnóstico18. En su inicio suele presentarse como una placa única, firme, indurada asintomática, de coloración violácea, roja-marronácea o rosada, de consistencia dura y adherida a la piel, pero no a planos profundos (figs. 1a y b)19. Con los años la placa puede mantenerse estable durante un largo periodo de tiempo, o crecer lentamente, o entrar en una fase de crecimiento rápido desarrollando múltiples nódulos, de ahí su nombre de protuberans (figs. 1c y d). No obstante, en series amplias se ha descrito que la mitad de los casos muestran ya una morfología protuberante desde su inicio20. Las lesiones en placa se han descrito en DFSP que simulan placas de morfea, otras deprimidas que parecen lesiones de atrofodermia y otras placas eritematosas o violáceas que recuerdan a un hemangioma. En el adulto la forma más frecuente de presentación es el de una placa de gran tamaño con múltiples nódulos en su superficie. En el niño se presentan más comúnmente en las formas no protuberans, como placa de morfea, y en los casos congénitos como placa de atrofodermia o tipo malformación vascular.

a: Dermatofibrosarcoma protuberans en placa localizado en la región clavicular tipo placa morfeiforme; b: dermatofibrosacoma protuberans en placa en la espalda tipo placa atrófica; c: dermatofibrosarcoma protuberans multinodular en la zona lumbar; d: Dermatofibrosarcoma protuberans tipo nodular en el hombro.
El tamaño de la lesión es variable y depende del periodo transcurrido hasta su diagnóstico que, en ocasiones, es muy tardío. Normalmente suele tener un tamaño de 2 a 5cm de diámetro, aunque se han descrito casos gigantes de un tamaño superior a 20cm10,13,15,16. La sospecha clínica del DFSP debe confirmarse mediante biopsia antes de realizar la cirugía.
Diagnóstico histológicoEs imprescindible para el diagnóstico realizar una biopsia que incluya tejido celular subcutáneo. En el estudio histológico se observa una tumoración mal delimitada que infiltra difusamente toda la dermis y se extiende al tejido celular subcutáneo (figs. 2 a, b y c), formada por una proliferación densa y uniforme de células fusiformes, monomorfas de núcleo alargado, con presencia de colágeno intercelular y de pequeños capilares. Las células fusiformes se disponen en fascículos cortos entrelazados, arremolinados, siguiendo un patrón que habitualmente se denomina estoriforme. En algunas zonas las células irradian desde un foco central acelular, fibroso, dando una imagen característica en rueda de carro17.

Histología típica de un dermatofibrosarcoma protuberans.a: Panorámica con hematoxilina eosina; b y c: infiltración dérmica e hipodérmica por el tumor; d: panorámica de la tinción intensamante positiva con CD34; e: detalle de las células fusiformes entre los adipocitos que muestran tinción intensa de CD34.
En el DFSP las células neoplásicas muestran poco pleomorfismo y la actividad mitósica es baja, habitualmente de menos de 2 mitosis por 10 campos a gran aumento. La densidad celular del DFSP es mayor en la zona central que en la periferia del tumor, mostrando esta última zona unos bordes infiltrativos en las dermis y en el tejido celular subcutáneo. El DFSP característicamente infiltra el tejido celular subcutáneo con proyecciones digitiformes en forma de tentáculos, que pueden llegar a gran distancia de la parte central del tumor. La expansión lateral y en profundidad de estos cordones irregulares de células fusiformes puede ser considerable, y en ocasiones asemejarse al tejido conectivo fibroso16. Este hecho puede ser la causa de recurrencias después de una resección que parecía adecuada.
Se han definido distintos subtipos clínico-patológicos de DFSP (tabla 4)21–28. La variante histológica con peor pronóstico es el DFSP con componente de fibrosarcoma (DFSP-FS), que se presenta en aproximadamente entre el 10% y el 20% de los casos29. Hay que sospechar esta transformación fibrosarcomatosa ante lesiones de gran tamaño, de rápido crecimiento y que infiltran el músculo15.
Es importante que el patólogo describa en el informe anatomopatológico las áreas de FS por su implicación pronóstica, puesto que la mayoría de los autores encuentran que las zonas del FS están relacionadas con una mayor tendencia a la recidiva y a las metástasis30,31. Las áreas del FS pueden ocupar desde un 5% hasta un 90% del tumor. El componente de FS se distingue por una proliferación de células fusiformes más densamente celular que las áreas de DFSP convencional, dispuestas en fascículos alargados, que interseccionan en varios ángulos, creando un patrón en espina de pescado. La transición entre los 2 componentes puede ser gradual o abrupta. El componente fibrosarcomatoso presenta una infiltración más compresiva. Asimismo, estas zonas fibrosarcomatosas muestran un mayor número de mitosis y atipia celular que el DFSP clásico.
Ante la sospecha histológica de un DFSP es recomendable realizar un estudio inmunohistoquímico dirigido a descartar otros tumores de características histológicas parecidas. El hallazgo inmunohistoquímico más característico del DFSP es la positividad para el anticuerpo CD34 con una expresión que manifiesta entre un 80% y un 100% de las células neoplásicas (figs. 2d y e)10,32. Sin embargo, la ausencia de este marcador no excluye su diagnóstico. De hecho, la expresión del CD34 puede ser débil o ausente en las zonas de FS, y sirve como marcador para reconocer estas áreas29,33. El CD34 también es muy útil tras la cirugía, para asegurar que los márgenes de extirpación están libres de tumoración, diferenciando entre células neoplásicas (CD34 positivas) y los fibroblastos de la dermis sana adyacente (CD34 negativos)34 y para distinguir entre neoplasia residual y tejido cicatricial en los casos de DFSP recidivantes35. No obstante, como con cualquier otro marcador inmunohistoquímico, la expresión de CD34 no es exclusiva de las células neoplásicas del DFSP, y se ha descrito también en células de otros tumores benignos y malignos (tumor fibroso solitario, hamartoma dendrocítico dérmico, lipoma de células fusiformes, angiosarcoma, fibroma esclerótico, sarcoma epitelioide, nevus del tejido conectivo fibroblástico).
Ante la sospecha clínica de DFSP, pero con una histología que no apoya este diagnóstico, es recomendable realizar una nueva biopsia36 (fig. 3).
Estudio molecular
El DFSP presenta una translocación genética característica en la que están implicados 2 genes: el gen del factor de crecimiento derivado de las plaquetas (PDGFB) (22q13.1) y el gen del colágeno i alpha 1 (COL1A1) (17q/22), cuya fusión origina un nuevo gen quimérico con capacidad transformante.
La presencia en el DFSP del gen de fusión COL1A1-PDGFB puede demostrarse en el tumor por la técnica de FISH o biología molecular aislando ARN del tumor y utilizando el procedimiento de la RT-PCR. El gen quimérico COL1A1-PDGFB está presente en el 90% de los DFSP33,37–39. Por tanto, la detección del gen de fusión COL1A1-PDGFB confirma el diagnóstico de DFSP, pero su ausencia no lo excluye.
El gen de fusión COL1A1-PDGFB se ha encontrado en todas las variantes clínico-patológicas, confirmando que todas ellas corresponden a una entidad tumoral única, si bien presentan diverso fenotipo microscópico33.
La determinación de la t(17;22) por FISH o RT-PCR no es necesaria para el diagnóstico de la mayoría de los casos de DFSP. Sin embargo, dado el carácter específico de esta translocación el estudio molecular es muy útil y recomendable cuando hay dudas diagnósticas y, en el contexto de enfermedad avanzada, para determinar qué pacientes pueden responder a los fármacos inhibidores de la tirosina quinasa. La técnica de FISH es más sensible y se debe recomendar como primera prueba diagnóstica. La RT-PCR se debe solicitar en casos de FISH no valorables o negativos.
EstadificaciónLos DFSP superficiales que no están clínicamente fijados a planos profundos no necesitan ningún estudio de imagen. Cuando se sospecha la afectación local de planos profundos (afectación de fascia o músculo) la resonancia magnética es el estudio preoperatorio de elección (fig. 3)40. Recomendamos esta prueba en tumores grandes con extensa afectación en profundidad, casos recidivados y en localización anatómica comprometida (cabeza y cuello)41.
Dada la baja frecuencia de metástasis (<3%), no es necesario ningún estudio de extensión, salvo el guiado por síntomas36,42. En pacientes con enfermedad recurrente y para DFSP con áreas de FS es conveniente realizar una TAC pulmonar. El pulmón es la localización más frecuente de las metástasis43, aunque también se han descrito metástasis en el cerebro, el hueso, abdominopélvicas y en el corazón.
Al igual que la mayoría de sarcomas, la diseminación del DFSP a ganglios linfáticos regionales es un hecho excepcional, siendo hasta 3 veces menos frecuentes que la visceral. De manera excepcional se han publicado diseminaciones mixtas (hematógena y linfática)44,45. En caso que se produzca la diseminación ganglionar o hematógena el pronóstico empeora drásticamente, con una supervivencia de menos de 2 años tras el diagnóstico de las metástasis16.
TratamientoTatamiento quirúrgicoLa extirpación quirúrgica completa es el tratamiento de elección para el DFSP. La infiltración grasa del DFSP a modo de proyecciones digitiformes le confiere una arquitectura muy asimétrica. Estas extensiones pueden no ser apreciables clínicamente y pueden incluso pasar desapercibidas en el estudio histológico convencional si no se examina la totalidad de los bordes laterales y profundos de la pieza quirúrgica46. Recientemente varios estudios comparan la cirugía convencional con amplios márgenes y la cirugía micrográfica de Mohs (CMM)47,48, demostrando que con la CMM se consigue un porcetaje de recurrencias mucho menor (30% cirugía convencional vs. 3% CMM), y por otro lado disminuye el defecto resultante tras la cirugía (10,7cm de media tras cirugía convencional vs. 8,8cm tras CMM) preservando una mayor cantidad de tejido sano47. En conclusión, actualmente se recomienda la CMM como tratamiento de elección (nivel de evidencia 1B) (fig. 3). La variante de cirugía de Mohs más aceptada es la denominada slow Mohs o Mohs diferido (estudio en parafina), pero también se han empleado otras variedades como la técnica de Breuninguer, complete circumferential peripheral and deep margin assessment (evaluación completa del margen circunferencial periférico y profundo) o histología en 3D en secciones en parafina36,49,50. El mínimo margen de escisión debe ser de 1cm llegando en profundidad hasta la fascia, sin ser necesario su extirpación en la primera etapa.
En los hospitales donde no es posible realizar CMM y no se pueda remitir a los pacientes es aconsejable realizar el procedimiento estándar con cirugía convencional, tomando un margen de resección de 2-3cm, llegando en profundidad hasta la fascia (fig. 3)51–55. En las lesiones recurrentes las localizaciones complicadas, como cabeza y cuello, o en los subtipos histológicos poco usuales o más agresivos (DFSP-FS) es imprescindible realizar CMM.
Independientemente de la técnica empleada la resección quirúrgica debe ser siempre meticulosamente planeada, dependiendo del tamaño del tumor, la localización y el subtipo histológico. Es muy importante asegurarse la exéresis completa del tumor antes de cualquier procedimiento de reconstrucción, especialmente si se va a realizar una plastia.
RadioterapiaNo hay ensayos clínicos respecto al papel complementario de la radioterapia. La radioterapia jamás exime de realizar una cirugía adecuada, y no está indicada tras la cirugía con márgenes libres42. La radioterapia ha sido empleada de manera anecdótica en casos donde la cirugía suponía una gran deformidad cosmética o funcional, o más frecuentemente, después del tratamiento quirúrgico con márgenes positivos56–59. Sin embargo, las series publicadas incluyen un escaso número de pacientes y poco tiempo de seguimiento, y la mayoría de estos trabajos están realizados antes de la introducción de la terapia con imatinib. En nuestra experiencia, ante un DFSP con márgenes positivos es imprescindible realizar CMM y no aplicar radioterapia. En nuestro servicio hemos tratado 240 DFSP con CMM, 3 casos recurrieron y se resolvieron con una nueva cirugía, no siendo necesaria la utilización de radioterapia en ningún caso. Además, en aquellos pacientes a los que se les ha administrado radioterapia es obligado revisar la zona periódicamente, pues se han descrito progresiones de DFSP convencionales a DFSP-FS tras este tratamiento60,61, y existe la posibilidad de inducir otro sarcoma en la zona irradiada. En conclusión, la radioterapia se debe reservar para casos realmente excepcionales, únicamente como tratamiento paliativo a nivel local en DFSP irresecables o en pacientes con DFSP metastásico.
Tratamiento sistémicoEl DFSP no es sensible a la quimioterapia convencional para sarcomas de partes blandas, por lo que debe no utilizarse al menos en primera línea42.
Está aprobado el uso del imatinib mesilato en Europa para el tratamiento de tumores primarios inoperables, enfermedad local recurrente inoperable y DFSP metastásico62,63. Es recomendable la detección del gen de fusión COLA1A-PDGFB antes de comenzar el tratamiento con imatinib, ya que este fármaco actúa por fijación competitiva en el receptor PDGF de las células tumorales, bloqueando su actividad tirosina quinasa. El imatinib parece beneficioso como tratamiento neoadyuvante en casos localmente avanzados con lesiones muy extensas y difíciles de intervenir, con la finalidad de disminuir el tamaño tumoral y facilitar la cirugía. Se han descrito respuestas parciales en el 50-80% de las series63–66. Es recomendable comenzar el tratamiento con 400mg día, pues ha demostrado la misma eficacia que las dosis más altas, y son mucho mejor toleradas. En caso de ausencia de respuesta subir la dosis a 600-800mg. La duración óptima del tratamiento neoadyuvante no está bien definida, pero el efecto máximo se consigue alrededor del 5.°-6.° mes. En los pacientes tratados con imatinib es difícil valorar en la histología de algunas zonas la presencia o ausencia del tumor, por lo que es recomendable realizar técnicas inmunohistoquímicas y de biología molecular. Nuevos fármacos inhibidores tirosina quinasa (sunitib, sorafenib y pazopanib) están siendo utilizados en casos de resistencia al imatinib67–69.
SeguimientoNo es necesario ningún estudio de extensión durante el seguimiento, salvo el guiado por los síntomas. Se debe realizar exámenes clínicos cada 6 meses durante 5 años para la detección precoz de posibles recidivas locales (fig. 3). Se aconseja, a partir de entonces, intervalos anuales hasta el décimo año después de la cirugía36.La mayoría de las recurrencias ocurren durante los primeros 3 años después de la cirugía, pero se han descrito recidivas mucho más tardías.
En algunas ocasiones las pruebas de imagen mediante RM local pueden ser beneficiosas en el seguimiento de DFSP recurrentes o DFSP-FS, o en aquellos casos en los que la cirugía haya sido compleja (>2 etapas CMM) o lesiones localizadas en la cabeza y el cuello.
ConclusiónLos sarcomas cutáneos representan una parte importante de la dermatología oncológica. El dermatólogo tiene un papel fundamental en el abordaje integral del paciente con DFSP.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


