



















Sarcomas comprise a broad group of tumors, many of whose biological behavior and aggressiveness differ from one type to another. The therapeutic approach is generally multidisciplinary and often complex. Developments in surgical and oncological dermatology during the last few decades have positioned dermatologists as specialists in the diagnosis and treatment of skin cancer. The aim of this article is to review the main soft tissue sarcomas that typically affect the skin. Dermatofibrosarcoma protuberans is a low-grade malignant sarcoma. It exhibits slow-growth, is locally invasive, and has low metastatic potential (<3%). Mohs micrographic surgery is the treatment of choice. The COL1A1-PDGFB translocation should be analyzed in cases of unclear diagnosis and when it is necessary to identify candidates for tyrosine kinase inhibitors. Imatinib is indicated for the treatment of locally advanced and metastatic dermatofibrosarcoma protuberans.
Los sarcomas constituyen un grupo amplio de tumores, muchos de ellos con comportamiento biológico y agresividad diferentes entre sí, que habitualmente requieren un tratamiento multidisciplinario, frecuentemente complejo. El desarrollo en las últimas décadas de la dermatología quirúrgica y oncológica ha permitido que los dermatólogos se conviertan en los especialistas responsables del diagnóstico y tratamiento del cáncer cutáneo. El propósito de este artículo es revisar los principales sarcomas de partes blandas de localización típicamente cutánea.
El dermatofibrosarcoma protuberans es un sarcoma de bajo grado de malignidad, con un crecimiento lento e infiltrativo localmente y escasa capacidad metastásica (<3%). El tratamiento de elección es la cirugía micrográfica de Mohs. Es recomendable solicitar el estudio de la translocación COL1A1-PDGFB cuando existen dudas diagnósticas, y para determinar qué pacientes pueden responder a los fármacos inhibidores de la tirosina quinasa. El imatinib está indicado en el dermatofibrosarcoma protuberans localmente avanzado y metastásico.
Sarcomas are a large, heterogeneous group of rare tumors that are typically treated using a multidisciplinary, often complex, approach. Developments in surgical and oncological dermatology in recent decades have positioned dermatologists as specialists in the diagnosis and treatment of skin cancer. This is a particularly pertinent consideration in the case of cutaneous soft tissue sarcomas. Sarcoma treatment is dealt with in numerous guidelines, but none of these specifically deal with sarcomas involving the skin. In addition, they have been drawn up by experts from specialities involved in the different stages of diagnosing and treating sarcomas (oncologists, pathologists, radiologists, traumatologists, and general and plastic surgeons), but they are missing input from dermatologists. This input is essential, as in many cases, the treatment of a retroperitoneal sarcoma cannot be extrapolated to that of a cutaneous sarcoma. The aim of this article is to provide clear recommendations, supported by the best possible clinical evidence, on the management of the main cutaneous sarcomas from a dermatological perspective to facilitate good clinical practice.
General BackgroundSarcomas are a large group of tumors that in many cases have varying biologic behavior and levels of aggressiveness. Soft tissue sarcomas are a heterogeneous group of rare tumors of mesenchymal origin. They account for less than 1% of all malignant tumors in adults and 12% of those in children.1,2 The vast majority (80%) arise in the soft tissues (including the skin), whilst the remainder originate in the bone, or, less frequently, the organs.
Soft tissue sarcomas have a broad histopathologic spectrum, possibly because the embryonic mesenchymal cells from which they are derived can differentiate into many types of tissue. The World Health Organization's classification of soft tissue sarcomas is based on the possible tissue origin of different types of tumors, including fibrosarcoma, angiosarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, and synovial sarcoma. Over 100 histologic subtypes are contemplated in this classification.1,2
The most common subtype of soft tissue sarcoma is undifferentiated pleomorphic sarcoma, followed by liposarcoma, leiomyosarcoma, and myxofibrosarcoma. The main cutaneous variants are dermatofibrosarcoma protuberans (DFSP) and Kaposi sarcoma.3
Soft tissue sarcomas are diagnosed and classified according to histologic patterns, immunohistochemical findings, and associated cytogenetic alterations.1,3,4 Histologic assessment continues to be the main diagnostic tool in sarcomas. Immunohistochemistry is a useful tool for exploring histogenetic origin, while molecular techniques such as fluorescence in situ hybridization (FISH) techniques, reverse transcription polymerase chain reaction (RT-PCR), and sequencing enable the identification of specific chromosomal translocations in many sarcomas.
Several grading and staging systems have been developed to guide prognosis. The 2 most widely used systems are the French Fédération National de Centres de Lutte Contre le Cancer (FNCLCC) grading system (Table 1) and the American Joint Committee on Cancer (AJCC) Staging Manual (Table 2).5–7 The FNCLCC system is based on histologic differentiating parameters, number of mitoses, and the presence or absence of necrosis, while the AJCC system takes into account tumor size and location (superficial or deep), lymph node involvement, metastasis, and histologic differentiation.8,9
Fédération National de Centres de Lutte Contre le Cancer (FNCLCC) Grading System for Soft Tissue Sarcomas.7
| FNCLCC Grading Parameters | |
|---|---|
| Parameter | Criterion |
| Tumor differentiation | |
| Score 1 | Sarcoma resembling normal adult mesenchymal tissue |
| Score 2 | Sarcomas for which histologic typing is certain |
| Score 3 | Embryonal and undifferentiated sarcomas: sarcomas of doubtful type |
| Mitotic count | |
| Score 1 | 0-9/10 high-power fields (HPFs) |
| Score 2 | 10-19/10HPFs |
| Score 3 | 20/10HPFs |
| Necrosis (microscopic) | |
| Score 1 | No necrosis |
| Score 2 | ≤ 50% tumor necrosis |
| Score 3 | >50% tumor necrosis |
| Histologic grade | |
| Grade 1 | Total score 2, 3 |
| Grade 2 | Total score 3, 4 |
| Grade 3 | Total score 5, 6 |
TNM Classification of Soft Tissue Sarcomas From the American Joint Committee on Cancer (AJCC) Staging Manual (7th Edition, 2010), 2016 version.5
| AJCC Definitions and Staging System |
|---|
| Primary tumor (T) |
| TX: Primary tumor cannot be assessed |
| T0: No evidence of primary tumor |
| T1: Tumor ≤ 5cm in greatest dimension |
| T1a: Superficial tumor: located above superficial fascia without invasion of the fascia |
| T1b: Deep tumor: located beneath the superficial fascia or superficial to the fascia but with invasion of or through the fascia |
| T2: Tumor with a maximum diameter ≥5cm |
| T2a: Superficial tumor |
| T2b: Deep tumor |
| Regional lymph nodes (N) |
| NX: Regional lymph nodes cannot be assessed |
| NO: No regional lymph node metastasis |
| N1: Regional lymph node metastasis |
| Distant metastasis (M) |
| Mx: Distant metastasis cannot be assessed |
| M0: No distant metastasis |
| M1: Distant metastasis |
| Histologic grade |
| GX: Grade cannot be assessed |
| G1: Well differentiated |
| G2: Moderately differentiated |
| G3: Poorly differentiated or undifferentiated |
| Stage | Grade | Primary Tumor | Regional Lymph Nodes | Metastasis |
|---|---|---|---|---|
| IA | G1 or GX | T1a or T1b | N0 | M0 |
| IB | G1 or GX | T2a or T2b | N0 | M0 |
| IIA | G2 or G3 | T1a or T1b | N0 | M0 |
| IIB | G2 | T2a or T2b | N0 | M0 |
| III | Any G | Any T | Any N | M0 |
| IV | Any G | Any T | Any N | M1 |
Hematogenous spread is more common than lymphatic spread in soft tissue sarcomas. Overall, lymph node involvement is uncommon, but it is relatively frequent in certain entities, such as rhabdomyosarcoma, synovial sarcoma, clear cell sarcoma, and epithelioid sarcoma. Tumors with a poor prognosis based on the FNCLCC grading system (large, deep, and high-grade tumors) are associated with metastasis, typically to the lungs; 10% of all patients with soft tissue sarcomas have metastasis on diagnosis.6 Most soft tissue sarcomas seen by dermatologists, however, have a low risk of hematogenous spread, with the exception of angiosarcoma and subcutaneous leiomyosarcoma (Table 3).6
Main Cutaneous Sarcomas Classified by Histologic Subtype and Approximate 5-Year Survival Rates.
| Histologic Subtype | Histologic Grade | Prognosis | ||
|---|---|---|---|---|
| I | II | III | 5-Year Survival | |
| Kaposi sarcoma | 60%-100% depending on immune status | |||
| Dermatofibrosarcoma protuberans | 97%-100% | |||
| Fibrosarcomatous dermatofibrosarcoma | 90%-95% | |||
| Congenital fibrosarcoma | 90%-100% | |||
| Leiomyosarcoma | 97% dermal 65% subcutaneous | |||
| Liposarcoma | 80% | |||
| Pleomorphic sarcoma | 80%-90% | |||
| Epithelioid sarcoma | 70% | |||
| Malignant peripheral nerve sheath tumor | 60% | |||
| Angiosarcoma | 35%-40% | |||










In this review, we examine soft tissue sarcomas with a typical cutaneous location, with a particular focus on dermatofibrosarcoma protuberans (DFSP), pleomorphic dermal sarcoma, leiomyosarcoma, angiosarcoma, and Kaposi sarcoma. We have excluded other sarcomas that can affect the skin, such as epithelioid sarcoma, malignant nerve sheath tumor, and liposarcoma, as they are rarely seen by dermatologists.
The purpose of this article is not to provide an exhaustive review of all possible cutaneous and/or superficial sarcomas, but rather to provide dermatologists with a general guide to diagnosing and treating the most common tumors seen.
Dermatofibrosarcoma ProtuberansIntroductionDFSP is a rare, slow-growing, and generally indolent cutaneous sarcoma that accounts for 5% of all sarcomas. Approximately 80% to 90% of DFSPs are low-grade tumors and fewer than 3% metastasize, although local recurrence is common due to the invasive nature of the tumor. Complete surgical excision is the treatment of choice. Five-year survival is very high (99%-100%).12
Epidemiology and Clinical DiagnosisDFSP accounts for less than 0.1% of all skin tumors.10 Its estimated incidence is between 0.8 and 5 cases per million inhabitants per year.11 It is more common in young adults (with onset occurring between the second and fifth decades of life), although it can appear at any age, from birth to old age.13,14 It appears to affect men and women equally. DFSP is seen in people of all racial backgrounds, but it is more common in black people, in particular Bednar tumor, which is a pigmented form of DFSP. The trunk is the most common site for DFSP, with approximately 50% to 60% of all tumors occurring in this region.10 The next most common sites are the proximal extremities (20%-30%) and the head and neck (10%-15%), particularly, the scalp, forehead, and supraclavicular fossa.15,16
DFSP is a solitary, multilobulated tumor of varying shapes and sizes. The lesion feels very firm on palpation, and it is fixed to overlying but not underlying tissue.17 The clinical appearance of DFSP depends on time since onset. It grows very slowly and there have even been reports of tumors being diagnosed 50 years after onset.18 It tends to present as a solitary, firm, asymptomatic, indurated plaque with a violaceous, reddish-brown, or pink appearance; it has a hard consistency and is fixed to the skin but not the deep layers (Fig. 1 A and B).19 The plaque can remain stable for long periods of time, grow slowly, or enter a phase of rapid growth characterized by the development of multiple nodules that gave rise to the term protuberans (Fig. 1 C and D). Data from large series, however, indicate that over 50% of tumors have a protruding morphology from the outset.20 DFSP lesions include morphea-like plaques, depressed lesions similar to those seen in atrophoderma, and erythematous or violaceous plaques resembling hemangioma. The most common presentation in adults is a large plaque with multiple superficial nodules. Children more commonly have nonprotruding lesions that resemble a morphea plaque, or in congenital cases, an atrophoderma plaque or vascular malformation.
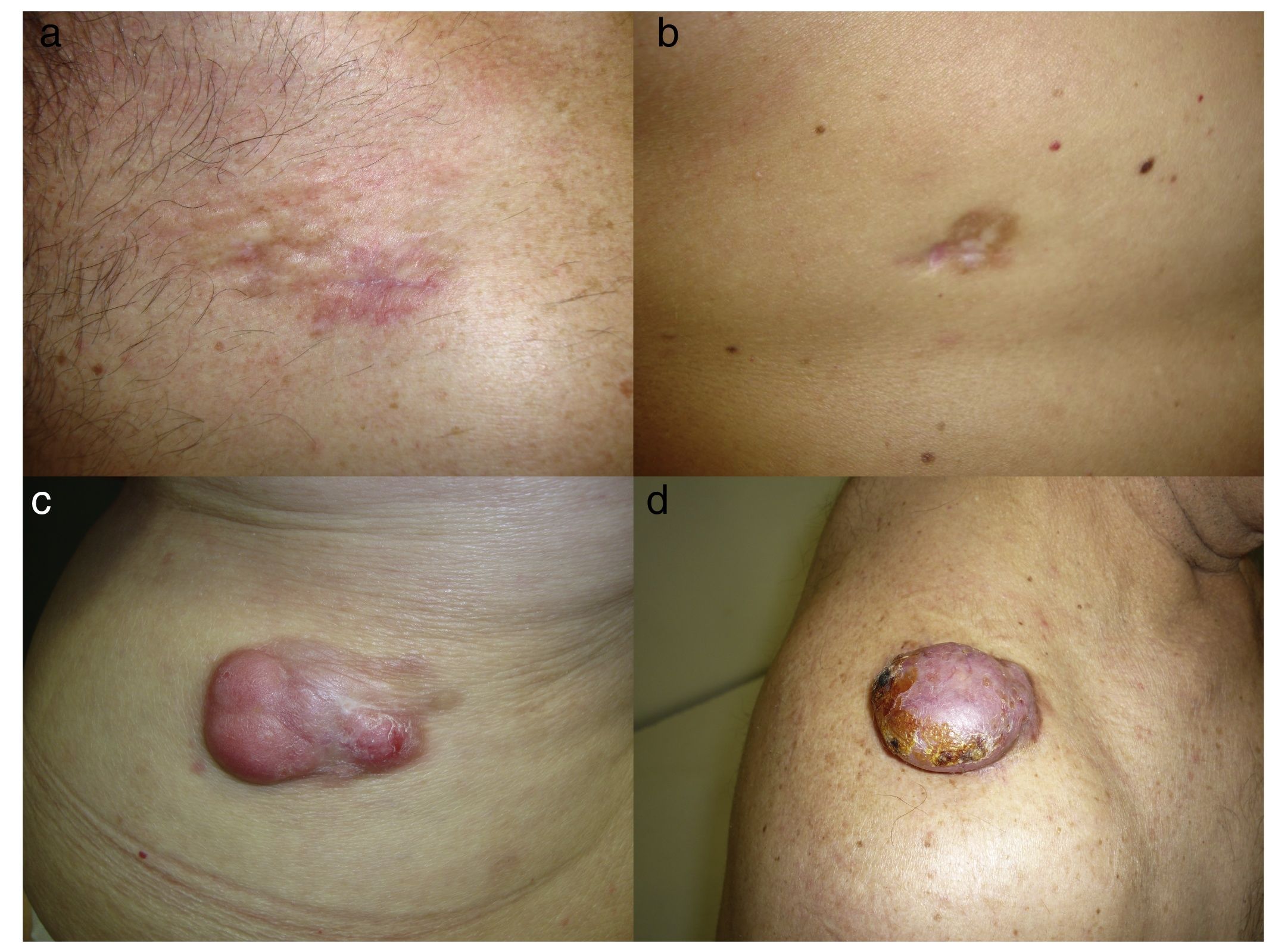
Lesion size is variable and depends on time to diagnosis, which can be considerable. Lesions normally measure 2 to 5cm in diameter, although there have been reports of giant lesions measuring over 20cm.10,13,15,16 A clinical suspicion of DFSP must be confirmed by biopsy prior to surgery.
Histologic DiagnosisThe biopsy specimen must include subcutaneous tissue. Histology shows a poorly delimited tumor invading the full thickness of the dermis and extending into the subcutaneous tissue (Fig. 2 A, B, and C). It is formed by a dense, uniform proliferation of monomorphous spindle cells with long nuclei, intercellular collagen, and small capillaries. Spindle cells have short interlaced, whorled fascicles that form a pattern typically described as storiform. In some areas, the cells irradiate from an acellular, fibrous focus, creating a characteristic cartwheel pattern.17
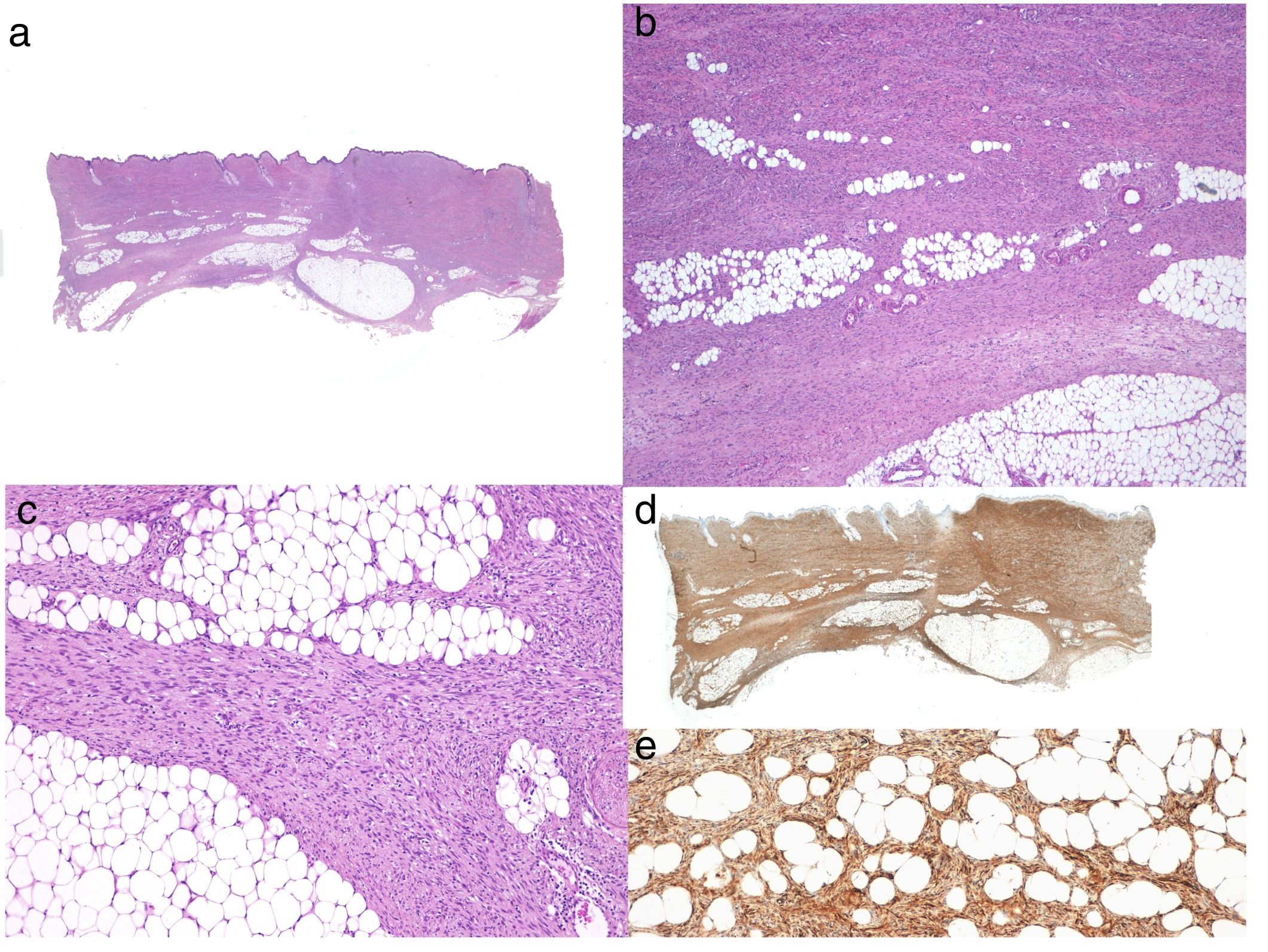
Neoplastic cells in DFSP are minimally pleomorphic and mitotic activity is low (generally < 2 mitotic figures per 10 high-power fields). Cells are denser in the center than at the periphery, where the edges of the tumor extend into the dermis and subcutaneous tissue. DFSP is characterized by tentacle-like projections that invade the subcutaneous tissue and can reach considerable distances from the center of the tumor. These irregular cords of spindle cells can extend quite far, both horizontally and vertically, and on occasions they resemble fibrous connective tissue,16 possibly explaining why certain tumors recur after what appeared to be an adequate resection.
Different clinicopathologic subtypes of DFSP have been defined (Table 4).21–28 The histologic subtype of DFSP with the worst prognosis is fibrosarcomatous DFSP (DFSP-FS), which accounts for approximately 10% to 20% of cases.29 DFSP-FS should be suspected in patients with large, fast-growing lesions that invade the muscle.15
Pathologists should describe fibrosarcomatous areas in the pathology report due to the prognostic implications, namely that DFSP-FS is associated with a greater tendency to recur and metastasize.30,31 Fibrosarcomatous areas can occupy between 5% and 90% of tumors. Fibrosarcomatous areas are characterized by a denser proliferation of spindle cells arranged in long fascicles that intersect at various angles, creating a fishbone pattern. The transition between fibromatous and conventional areas may be gradual or abrupt. Invasion by the fibrosarcomatous component of DFSP is characterized by greater compression. This component also has a higher rate of mitosis and cellular atypia than classic DFSP.
When there is histologic evidence of DFSP, an immunohistochemical study should be performed to rule out other tumors with similar features. The most characteristic immunohistochemical finding in DFSP is CD34 positivity, which is seen in 80% to 100% of neoplastic cells (Fig. 2 D and E).10,32 Absence of this marker, however, does not necessarily exclude a diagnosis. In fact, CD34 expression may be weak or absent in fibrosarcomatous areas; this marker can be used to identify the presence of a fibrosarcomatous component.29,33 CD34 is also very useful for checking for tumor-free margins after surgery, differentiating between neoplastic cells (CD34+) and fibroblasts in healthy adjacent skin (CD34-),34 and distinguishing between residual tumor and tumor scarring in cases of recurrent DFSP.35 Nonetheless, like all immunohistochemical markers, CD34 is not only found in neoplastic DFSP cells, but has also been described in other benign and malignant tumors, including solitary fibrous tumor, dermal dendrocyte hamartoma, spindle cell lipoma, angiosarcoma, sclerotic fibroma, epithelioid sarcoma, and fibroblastic connective tissue nevus.
A repeat biopsy is recommended when a clinically suspicious DFSP is not confirmed histologically36 (Fig. 3).
Molecular Profiling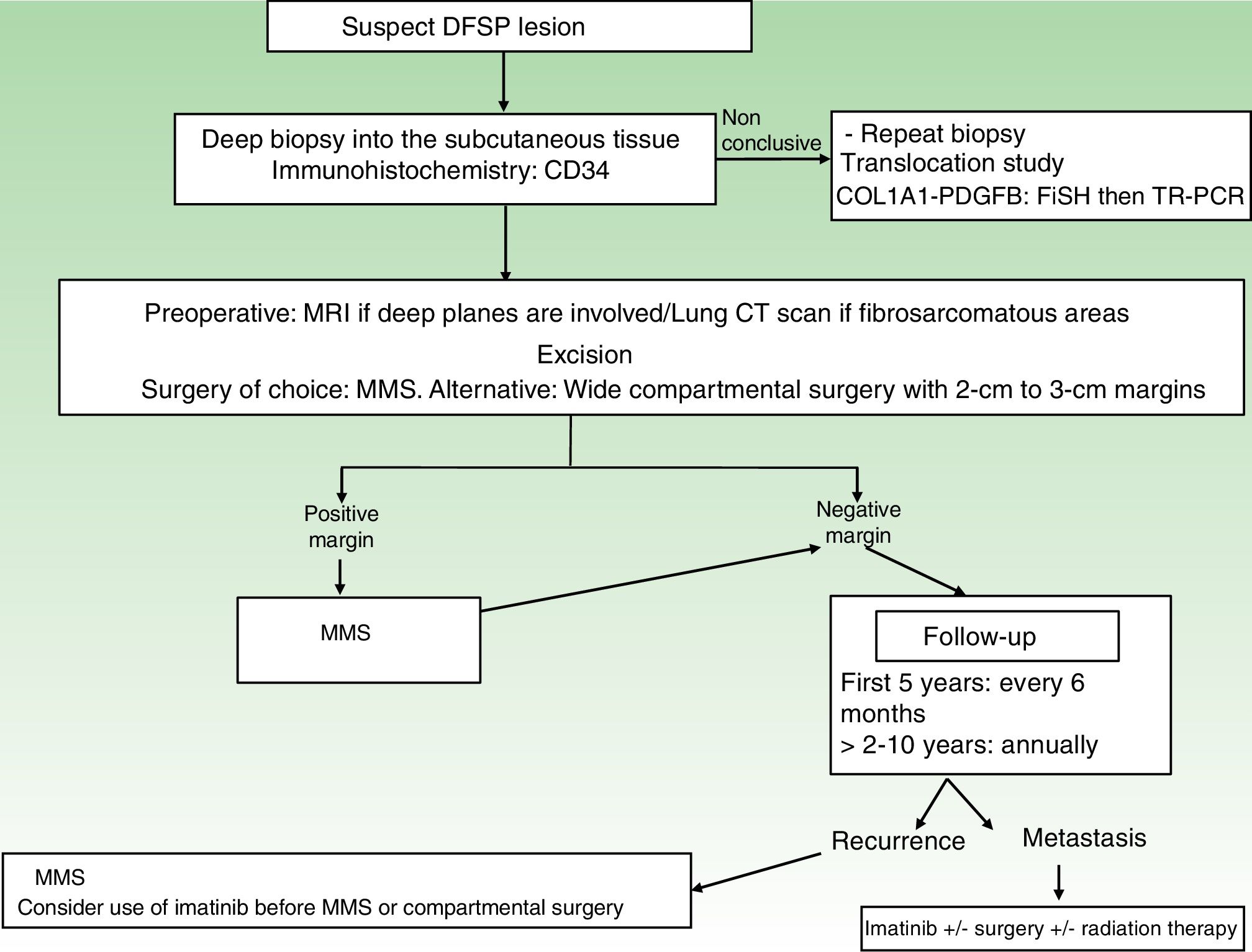
DFSP has a characteristic translocation of genetic material involving 2 genes: the platelet-derived growth factor subunit B gene (PDGFB) and the collagen type 1 alpha 1 gene (COL1A1), whose fusion gives rise to a new chimeric gene with transformative capacity.
The COL1A1-PDGFB fusion gene in DFSP can be detected by isolating RNA from the tumor using FISH or molecular biology techniques or by performing RT-PCR. This chimeric gene is present in 90% of DFSPs.33,37–39 While detection of the gene thus confirms a diagnosis, failure to do so does not rule one out.
The COL1A1-PDGFB fusion gene has been found in all clinicopathologic variants of DFSP, confirming that all these entities constitute a single tumor entity, albeit with varying histologic phenotypes.33
Identification of the t(17;22) translocation by FISH or RT-PCR is not necessary for diagnosis in most cases. However, considering the specific nature of this translocation, molecular assays are very useful, and advisable, when there are diagnostic doubts or in cases of advanced disease to identify candidates for tyrosine kinase inhibitors. FISH is more sensitive and should be performed first. RT-PCR should be ordered in cases of unassessable or negative FISH tests.
StagingSuperficial DFSPs that are not clinically fixed to the deeper planes do not require imaging studies. When local involvement of the deep planes (fascia or muscle) is suspected, magnetic resonance imaging (MRI) is the preoperative test of choice (Fig. 3).40 We recommend using MRI to investigate large tumors extending into the deeper layers, recurrent tumors, and tumors in complicated anatomic locations (head and neck).41
As metastasis is rare (<3% of cases), staging studies are not required, unless prompted by symptoms.36,42 A computed tomography scan of the lungs is advisable in patients with recurrent disease and DFSP-FS. The lungs are the most common site for metastasis from DFSP,43 although the tumor has also been known to spread to the brain, bone, abdominal-pelvic region, and heart.
Like most sarcomas, DFSP rarely spreads to the regional lymph nodes. In fact, regional lymph node metastasis is 3 times less common than visceral metastasis. There have been isolated reports of mixed patterns of spread (hematogenous and lymphatic).44,45 Prognosis is dramatically worse for patients with metastasis (survival ≤2 years following diagnosis of disease spread).16
TreatmentSurgical TreatmentComplete surgical excision is the treatment of choice for DFSP. The growth of DFSP, with tentacles extending into fatty tissue, is highly asymmetric. These extensions may not be clinically evident and can even go unnoticed in conventional histology if the lateral and deep margins of the surgical specimen are not examined in their entirety.46 Several recent studies that have compared conventional wide local excision (WLE) and Mohs micrographic surgery (MMS)47,48 have demonstrated that MMS is associated with a much lower recurrence rate than WLE (3% vs 30%), a smaller surgical defect (8.8cm vs 10.7cm), and greater sparing of healthy tissue.47 In conclusion, MMS is currently the surgical treatment of choice for DFSP (supported by level of evidence 1B) (Fig. 3). The most widely accepted variant of MMS is slow Mohs (paraffin study), but other techniques have also been used, including the Breuninguer technique, complete circumferential assessment, and 3D histology assessment of paraffin-embedded sections.36,49,50 The minimum excision depth should be 1cm, i.e., it should extend down to the fascia, which does not need to be removed in the first step.
In hospitals where it is not possible to perform MMS or refer patients for this option, conventional surgery with deep margins of 2 to 3cm, extending into the fascia, is recommended (Fig. 3).51–55 MMS is essential for recurrent lesions, lesions in complicated locations, such as the head or neck, and uncommon or more aggressive histologic subtypes (DFSP-FS).
Regardless of the technique employed, surgical resection must be carefully planned to take into account tumor size, location, and histologic subtype. It is very important to check that all traces of tumor have been removed prior to reconstruction and particularly before flap-repair procedures.
Radiation TherapyThe role of radiation therapy as an adjuvant treatment in DFSP has not been analyzed in a clinical trial to date. Radiation therapy is never a substitute for adequate surgical excision, and is not indicated as a postoperative treatment for patients with tumor-free margins.42 There have been isolated reports of radiation therapy being used when surgery would have led to major cosmetic or functional defects, and, more frequently, following surgical treatment with positive margins.56–59 Nevertheless, the series published to date have analyzed very few patients and the follow-up times have been very short. In addition, most of these studies were performed before the introduction of imatinib therapy. In our experience, MMS is essential in patients with DFSP with positive margins and radiation therapy should not be administered. In our department, we have treated 240 DFSPs with MMS; 3 of the tumors recurred and were resolved with repeat surgery; radiation therapy was not necessary in any of the cases. Regular monitoring is essential in patients administered radiation therapy, as there have been reports of conventional DFSP progressing to DFSP-FS following this treatment.60,61 In addition, radiation therapy could induce another sarcoma in the irradiated area. To conclude, radiation therapy should be reserved for truly exceptional cases, such as unresectable tumors (palliative local therapy) and metastatic DFSP.
Systemic TreatmentDFSP does not respond to conventional chemotherapy used to treat soft tissue sarcomas and should therefore not be used, at least as a first-line option.42
In Europe, imatinib mesylate is approved for the treatment of inoperable primary tumors, inoperable local recurrent tumors, and metastatic DFSP.62,63 Tests to detect the fusion gene COLA1A-PDGFB are advisable prior to treatment with imatinib, as this drug acts by competitive binding to platelet-derived growth factor receptors on tumor cells, blocking their tyrosine kinase activity. Imatinib appears to be useful as a neoadjuvant treatment for reducing tumor size and facilitating surgery in locally advanced cases with extensive, difficult-to-access lesions. Partial responses of 50% to 80% have been reported by the series published to date.63–66 The recommended starting dose is 400mg/d, as this dose offers the same efficacy as higher doses but is much better tolerated. The dose can be increased to 600 to 800mg in the absence of response. Optimal duration of neoadjuvant treatment has not been well defined, but peak effects are achieved around month 5 or 6. Immunohistochemical and molecular profiling studies are recommended in patients treated with imatinib, as in some areas of the tumor, it can be difficult to determine the presence or absence of neoplastic cells. New tyrosine kinase inhibitors (sunitib, sorafenib, and pazopanib) are being used in patients resistant to imatinib.67–69
Follow-upStaging studies are not needed during follow-up, unless prompted by symptoms. Clinical examinations are necessary every 6 months over 5 years to enable early detection of local recurrences (Fig. 3). The recommendation thus is to schedule annual follow-up visits for 10 years following surgery.36 Most recurrences occur within 3 years of surgery, although much later recurrences have been described.
On occasions, local MRI can be useful for monitoring recurrent DFSP or DFSP-FS, complicated surgical cases (those requiring >2 MMS steps), and tumors located on the head and neck.
ConclusionsCutaneous sarcomas are an important entity in oncological dermatology. Dermatologists play an important role in the overall management of patients with DFSP.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Llombart B, Serra C, Requena C, Alsina M, Morgado-Carrasco D, Través V, et al. Sarcomas cutáneos: directrices para el diagnóstico y tratamiento. Dermatofibrosarcoma protuberans. Actas Dermosifiliogr. 2018;109:868–877.


