


El término de Sarcoma de Ewing procede de James Ewing (1866-1943), que fue el primero en describir el tumor como un endotelioma óseo maligno, estableciendo que la enfermedad era diferente del linfoma1,2. Angervall y Enzinger identificaron que el tumor podía producirse en el tejido blando profundo y pocas veces en la piel3. Posteriormente, los estudios citogenéticos confirmaron que estos tumores pertenecen a la misma familia que el sarcoma de Ewing que surge del hueso, que comúnmente reciben el nombre de tumores de la familia del sarcoma de Ewing4. Hasta el 2018 únicamente se han descrito en la literatura 78 casos. La mayoría de los pacientes son mujeres (2:1), de raza caucásica, en la segunda década de la vida (edad media entre los 16-17 años). Se ha demostrado la translocación del gen EWSR1 en el cromosoma 22q12 así como la hibridación fluorescente in situ del gen EWSR1 con una rotura positiva para la translocación de EWSR11,2,5.
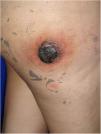
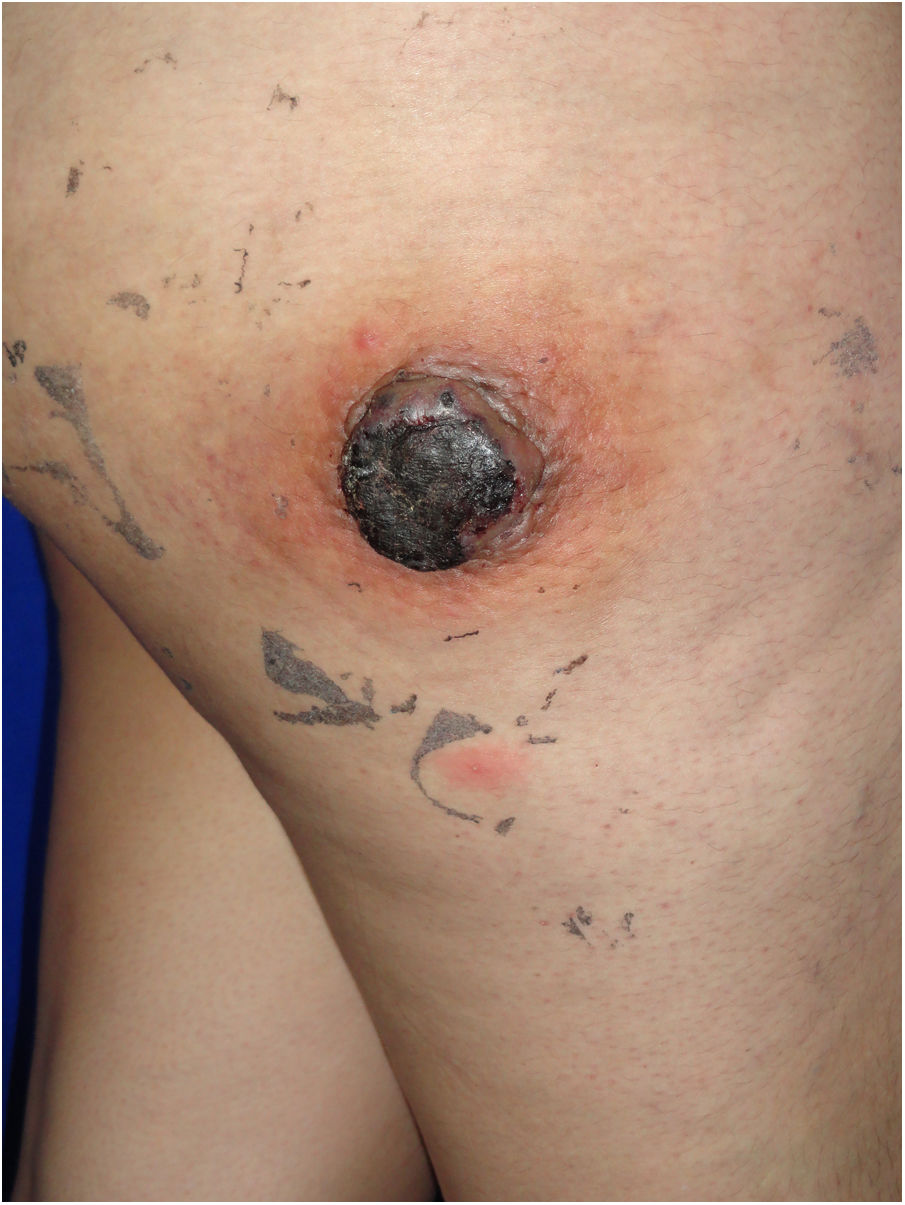
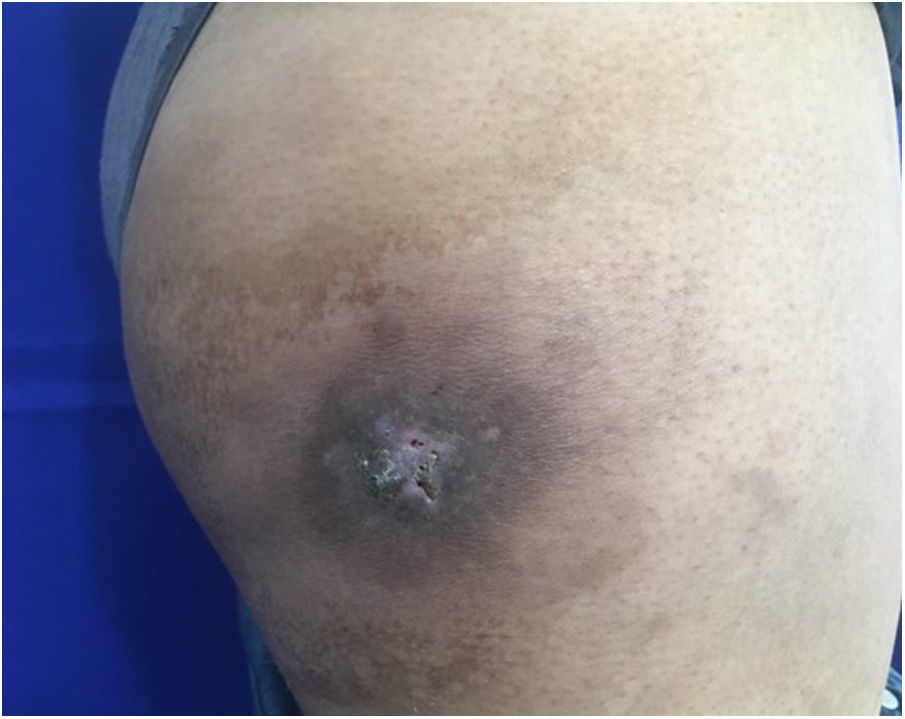
Caso clínicoUna mujer de 25 años acudió a consulta, tras 22 días de su parto, por la aparición, 7 meses antes, de una masa de rápido crecimiento en el muslo derecho. La paciente refería fatiga y dolor asociado, además de sangrado de la masa. El examen físico reveló una lesión tumoral eritematosa, firme, de 5cm de diámetro, con costras hemáticas en la superficie, rodeada por un halo eritematoso de piel de naranja (fig. 1).
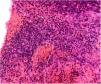
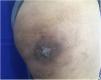
Se realizó una biopsia con sacabocados que mostró un tumor de células pequeñas basófilas, con poco citoplasma, cromatina y núcleo prominente, y células dispuestas en capas a lo largo del grosor de la dermis sin compromiso epidérmico (fig. 2). El estudio inmunohistoquímico fue positivo para CD99, PKC, y vimentina, y negativo para actina, desmina, S100, cromogranina, CD20, C3 y CK20. La TAC torácica reveló un nódulo sólido de naturaleza no cálcica de 5mm en la base del pulmón izquierdo, sospechoso de metástasis, además de paquipleuritis izquierda. La TAC abdominal mostró hepatomegalia a expensas del lóbulo derecho, sin dilatación del conducto biliar, no se observaron masas sólidas ni quísticas en el parénquima; además, se detectó esplenomegalia con una imagen heterogénea hipodensa de tamaño no mayor a 25mm, también sospechosa de metástasis. Se diagnosticó un tumor neuroectodérmico primitivo tipo sarcoma primario cutáneo de Ewing. La paciente fue referida a un hospital donde recibió un ciclo de inducción con vincristina y doxorrubicina, logrando una reducción del 50% de la masa. Este fue seguido de 2 ciclos de vincristina ya que se agotaron los suministros. El sangrado se presentó con el tercer ciclo, por lo que se indicó radioterapia (acelerador lineal). Seis meses más tarde, se obtuvo una reducción del 90% de la lesión cutánea (fig. 3). Desgraciadamente la paciente falleció 2 semanas después de la última cita en dermatología.
Discusión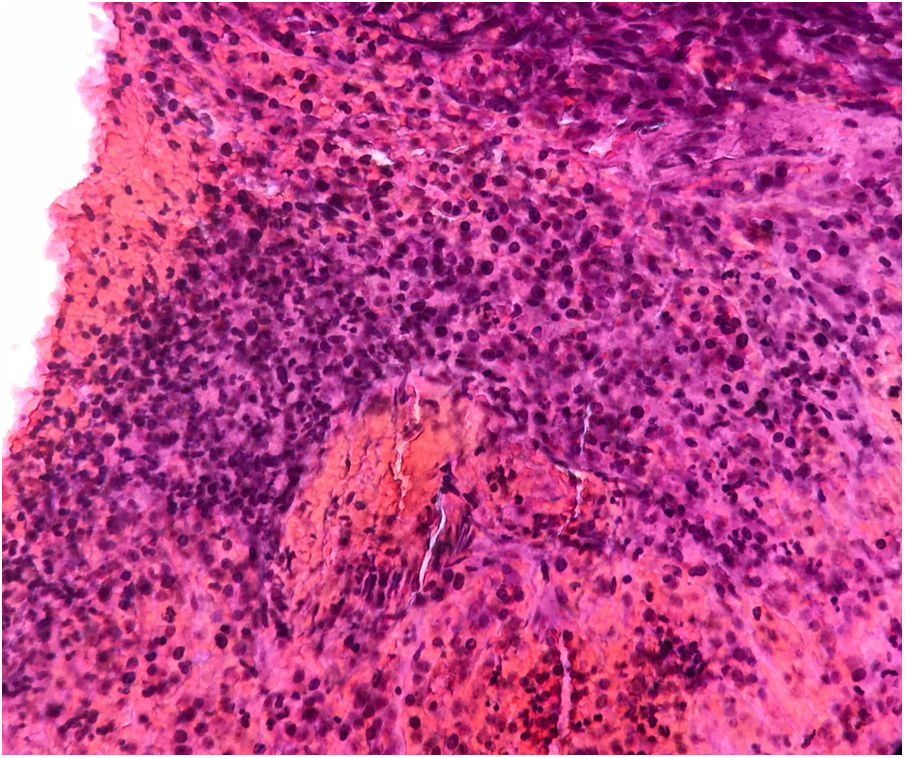
Tradicionalmente, la familia de tumores del sarcoma de Ewing incluye 3 entidades significativas: el sarcoma de Ewing, el tumor de Askin de la pared torácica y el PNET1,4.
El sarcoma de Ewing es un tumor neuroectodérmico primitivo que raramente se presenta en piel y tejido subcutáneo. El 85% de los tumores presentan fusión del gen EWSR1 con un gen transformador del virus de la eritroblastosis (FLI1); la fusión del exón 7 de EWSR1 al exón 6 de la translocación FLI1 es específica del sarcoma de Ewing1. En general, es más conocido como un tumor óseo primario con una mayor incidencia en niños y adolescentes, siendo en este grupo de edad el segundo tumor óseo primario más común2,4. El sarcoma de Ewing es poco frecuente, localizándose principalmente en los tejidos blandos profundos de la región paraespinal, la pared torácica o las extremidades inferiores. Los casos más superficiales, denominados cutáneos, son esporádicos y la gran mayoría han sido descritos como una única masa pequeña1,2.
Histológicamente, estos sarcomas están constituidos principalmente por células tumorales pequeñas y redondas que, por lo general, expresan CD99 y positividad débil para marcadores de sinaptofisina.3,5–8. Algunos tumores pediátricos de células pequeñas y redondas con afectación cutánea pueden descartarse fácilmente mediante inmunohistoquímica. El rabdomiosarcoma puede exhibir positividad para CD99 y también tiñe marcadores musculares, como la desmina, miogenina y myo-D1, lo que no sucede con los tumores de la familia del sarcoma de Ewing4,5,8. El linfoma linfoblástico puede afectar a la piel y frecuentemente es positivo para CD99 y también para TdT, mientras que el tumor de la familia del sarcoma de Ewing es siempre negativo para este último2,4,5,7.
El sarcoma cutáneo primario de Ewing es una entidad rara, que se trata de la misma forma que el sarcoma óseo de Ewing, mediante cirugía extensa, radioterapia y quimioterapia multifármaco1,7. La enfermedad cutánea presenta un curso lento y un pronóstico favorable, con una tasa de supervivencia a los 10 años del 91%, a diferencia de lo que sucede con el sarcoma de Ewing óseo o de tejidos blandos, que presentan un peor pronóstico. El comportamiento menos agresivo del cuadro clínico cutáneo probablemente es debido a la localización superficial, a ser tumores más pequeños y a tener un acceso fácil; todo ello permite su detección, bien mediante un examen clínico rutinario, bien por autoexploración del paciente. Esto hace posible llegar a un diagnóstico precoz y lograr una resección quirúrgica completa, lo que evita la aparición de metástasis.1,2,4,7 Si la enfermedad se detecta a tiempo y se trata con escisión local y quimioterapia sistémica, el resultado es favorable. Actualmente no existe literatura científica que demuestre una relación directa y causal entre el sarcoma de Ewing cutáneo primario y el embarazo. Desafortunadamente, como es el caso que presentamos, el 11% de los casos de sarcoma de Ewing cutáneo primario presentan enfermedad metastásica en el momento del diagnóstico4,5,8.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.


