




Dystrophic epidermolysis bullosa is a rare inherited disease caused by mutations in the COL7A1 gene. Its recessive variant (recessive dystrophic epidermolysis bullosa) is characterized by the absence or considerably reduced expression of type VII collagen, which leads to marked fragility of the skin and mucous membranes and subsequent blister formation, whether spontaneously or following minimal injury. There have been very few reports of this disease in pregnant women.
We present 2 cases of pregnant women with recessive dystrophic epidermolysis bullosa managed in our High-Risk Pregnancy Unit at Hospital Universitario La Paz, Madrid, Spain. Both patients underwent full-term cesarean delivery, with no further complications for mother or child.
Although recessive dystrophic epidermolysis bullosa increases the risk of maternal complications, a patient is not advised against pregnancy. With adequate monitoring, these patients can fulfil their desire to become mothers.
La epidermólisis bullosa distrófica es una enfermedad hereditaria rara debida a mutaciones del gen COL7A1. Su variante recesiva (EBDR) se caracteriza por una marcada disminución o ausencia completa de colágeno tipo VII (C7), que da lugar a una marcada fragilidad de la piel y las mucosas, desencadenando la formación de ampollas de forma espontánea o en respuesta a mínimos traumatismos. Son muy pocos los casos descritos en la literatura de esta enfermedad en embarazadas.
Exponemos 2 casos de gestantes, ambas afectadas de EBDR, y su manejo en nuestra Unidad de Obstetricia de Alto Riesgo del Hospital Universitario La Paz. En ambos casos se realizó una cesárea a término, finalizando la gestación sin complicaciones mayores para la madre o el feto.
A pesar de relacionarse con un mayor número de complicaciones maternas, la EBDR no representa una contraindicación para la gestación, y con un control adecuado, estas pacientes pueden ver su deseo genésico cumplido.

Epidermolysis bullosa (EB) is a heterogeneous group of hereditary diseases characterized by marked fragility of the skin and mucous membranes and subsequent blister formation, either spontaneously or following minimal injury. EB is a rare disease that can affect people of any ethnic background; men and women are equally affected.1
EB is classified in 4 major types according to the plane of cleavage in the skin: epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB), and Kindler syndrome (KS).2
DEB is caused by mutations in the COL7A1 gene, which encodes type VII collagen. Type VII collagen is the main component in the anchoring fibrils that link the epithelium to the connective tissue and is therefore essential for maintaining the integrity of the skin and mucous membranes. Severe generalized recessive dystrophic epidermolysis bullosa (RDEB) is the most severe form of EB. It is characterized by a marked decrease in—or complete absence of—type VII collagen, which results in mucosal and cutaneous erosions, chronic wounds, and aggressive squamous cell carcinomas.
There have been very few reports of RDEB in pregnant women due to the significant morbidity associated with these cases. We present 2 cases of pregnant women with RDEB that were managed in our High-Risk Pregnancy Unit at Hospital Universitario La Paz, Madrid, Spain.
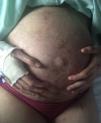
Case DescriptionsCase 1The patient was a 40-year-old woman with severe generalized RDEB. Physical examination revealed erosions and blisters on most of the body and pseudosyndactyly of the hands and feet. As an extracutaneous complication, the patient developed esophageal stenosis, which made it difficult to swallow solids. The patient did not have a gastrostomy tube. Her partner was affected by retinosis pigmentaria. Direct sequencing of the COL7A1 gene revealed 2 heterozygous mutations in exons 34 and 80. Pregnancy was achieved by in vitro fertilization with the couple's own oocytes and semen because vaginal stenosis prevented sexual relations. Amniocentesis at week 15 of pregnancy ruled out fetal RDEB. As complications of pregnancy, the patient developed hypothyroidism, which was treated with levothyroxine (100μg/d), iron-deficiency anemia, which was treated with intravenous iron (oral administration was impossible due to the patient's difficulty swallowing), and gestational diabetes, which was controlled with diet. The patient also experienced several episodes of secondary infection of the skin lesions with Serratia organisms and Staphylococcus aureus, which responded to antibiotic therapy with amoxicillin–clavulanic acid and cefuroxime (Fig. 1).
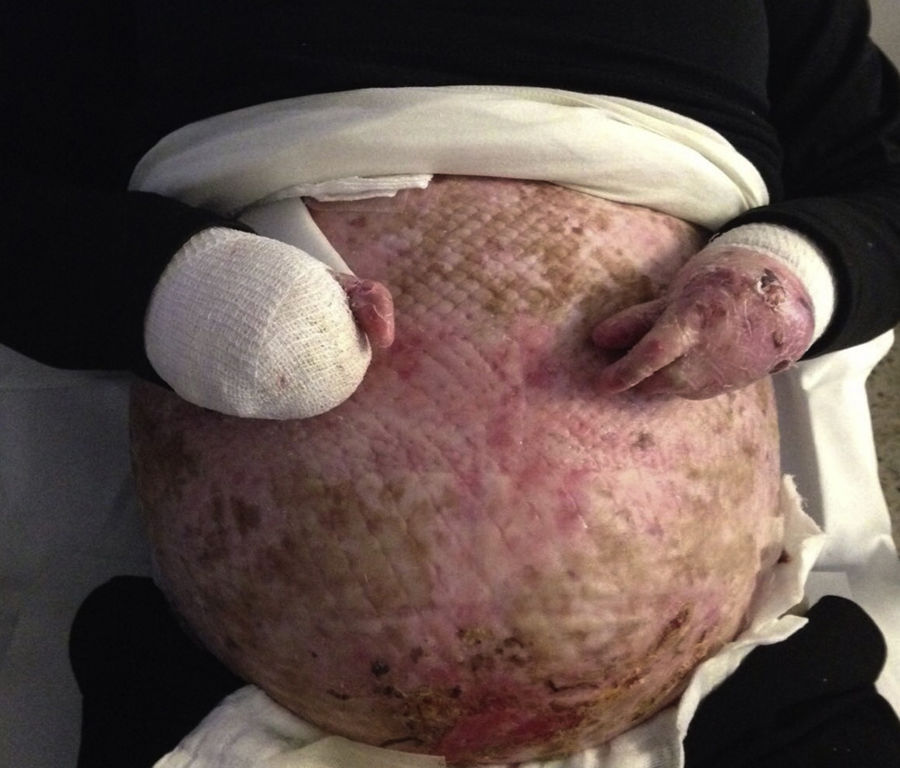
Follow-up ultrasound at 32 weeks found that the fetus was small for gestational age, with no hemodynamic effects. Cesarean section was performed at week 37+4 days due to vaginal stenosis; the infant girl weighed 2200g at birth and was not affected by RDEB. The postsurgical period was without incident and reepithelialization of the surgical wound was achieved after a few weeks. The patient was advised against breastfeeding due to periareolar involvement.
Case 2The patient was a 25-year-old woman with moderate cutaneous involvement and esophageal dysphagia. Genetic analysis identified 2 heterozygous mutations: the maternal mutation p.R2008C, found on exon 73 of the COL7A1 gene, and the paternal mutation p.R1730X, found on exon 58 of the COL7A1 gene. The patient's medical history included a previous pregnancy monitored at another hospital with threat of premature delivery at week 30 and cesarean section at week 34. The current pregnancy was spontaneous. The patient developed iron-deficiency anemia and vitamin D deficiency with poor oral tolerance in the first trimester, which required treatment with vitamin D supplement packets and 2 hospital stays for intravenous iron therapy. Cesarean section was performed at week 36+2 days due to vaginal stenosis; the infant girl weighed 2980g at birth. The postsurgical period was without incident and the patient was able to start breastfeeding immediately after delivery (Fig. 2).
Discussion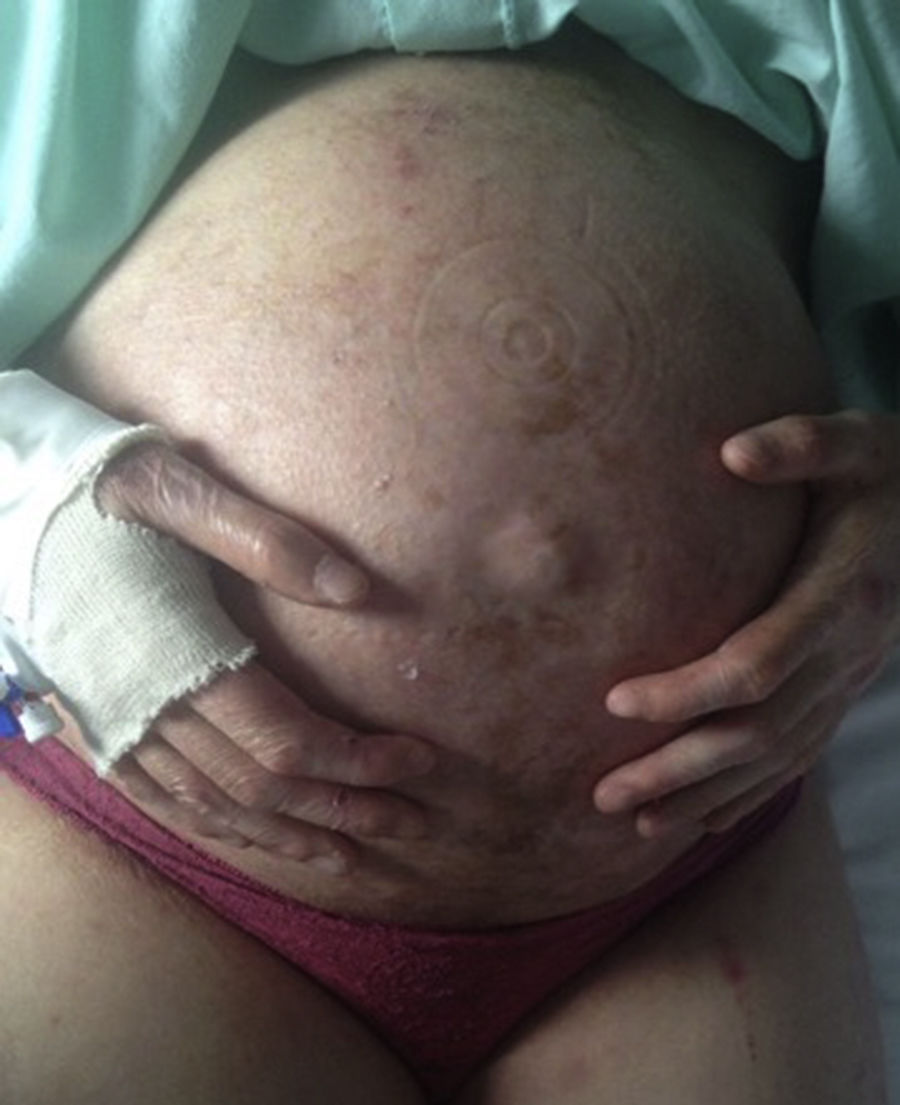
The diagnostic process for EB begins with an adequate medical history that includes relevant family history and a complete physical examination. The next step is to make an initial classification of the condition as one of the 4 main EB types by using immunofluorescence antigen mapping to identify the plane of cleavage. Once the affected protein is identified, genetic analysis can be carried out by sequencing the candidate gene.
In both of our patients, mutations were found in the COL7A1 gene; it was therefore possible to perform amniocentesis to reach a prenatal diagnosis in one of the women.
Genetic counseling and guidance are fundamental in these patients. Because RDEB is a recessive disease, prenatal or preimplantation diagnosis is only necessary if the patient's partner is suspected of being a carrier.
No definitive cure for EB currently exists. Treatment of this disease is symptomatic and supportive, focusing primarily on preventing complications. Therapeutic approaches to RDEB aim to correct the deficiency or absence of specific anchor proteins in the dermoepidermal junction, with a particular focus on restoring expression of type VII collagen. Various protein-based, cellular, and genetic therapies are currently being investigated.3–5
Although the literature on this subject is limited, the available evidence seems to indicate that RDEB increases the risk of maternal complications but does not contraindicate pregnancy.6–11 In our experience, with strict monitoring made it possible to bring both of our patients’ pregnancies to term without major complications for mother or child. Early treatment of iron deficiency, vitamin deficiency, and skin infections are the cornerstone of pregnancy management in these patients. Although vaginal births without incident have been reported in pregnant women with RDEB, the type of delivery should be determined on an individual basis in accordance with the conditions of each patient.7,9,11 Scheduled cesarean section was performed in both of our patients due to vaginal stenosis. However, for patients without vaginal involvement, and in the absence of comorbidities that would contraindicate vaginal delivery (multiple previous cesarean sections, placenta previa, etc.), this could be an option to consider towards the end of pregnancy.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Boria F, Maseda R, Martín-Cameán M, De la Calle M, de Lucas R. Epidermólisis bullosa distrófica recesiva y embarazo. Actas Dermosifiliogr. 2019;110:50–52.


