


INTRODUCCIÓN
Aunque el rabdomiosarcoma es el tumor maligno más frecuente de partes blandas en niños1 , la localización vulvar y la presentación al nacimiento son excep-cionales2-7 . Los tumores vulvares son un motivo de consulta infrecuente en Dermatología y más aún en pacientes pediátricos. El reconocimiento del aspecto característico de este tumor permitirá un diagnóstico precoz y una actuación terapéutica temprana.
DESCRIPCIÓN DEL CASO
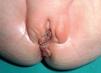
Una niña de una semana de vida fue traída a la consulta de dermatología por presentar una tumoración vulvar que había crecido rápidamente desde el nacimiento. La niña no presentaba ningún antecedente de interés, y había nacido a término fruto de un embarazo y un parto sin incidencias. A la exploración se observó la presencia de lesiones tuberosas de color rojo oscuro y con apariencia en “racimo de uvas” que ocupaban la mucosa de la vulva y se extendían hacia la piel de los labios mayores (fig. 1).

Fig. 1.—Masa excrecente en vulva con aspecto en racimo de uvas.
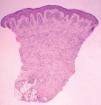
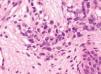
Se realizó una biopsia de la lesión del labio mayor. En el estudio histológico llamaba la atención la presencia de una condensación celular en banda subepidérmica (capa de cámbium) (fig. 2). Se apreció una infiltración cutánea por una neoplasia mesenquimatosa constituida por elementos rabdomioblásticos atípicos redondeados o fusiformes (fig. 3). Las células se disponían de forma vagamente estoriforme sobre una matriz mixoide. Con técnicas de inmunohistoquímica se evidenció positividad para vimentina, MyoD1 y marcadores de diferenciación muscular (actina HHF35, desmina y mioglobina). Las células neoplásicas eran negativas para queratina AE1/AE3, EMA, HMB45, S100, enolasa neuronal específica, CD99, CD34 y CD45.
En la resonancia magnética (RM) se observó afectación de la vulva sin evidencia de extensión a la vagina o a planos musculares. La tomografía computarizada (TC) toracoabdominal no mostró evidencias de enfermedad a distancia. Con el diagnóstico de rabdomiosarcoma botrioides se inició tratamiento con quimioterapia según el protocolo MMT-95/01 de la Sociedad Internacional de Oncología Pediátrica. Posteriormente se realizó cirugía parcial y se continuó con la quimioterapia, consiguiéndose una primera remisión completa 9 meses después del diagnóstico. Diez meses después, la paciente presentó una recidiva en la zona perianal, que se confirmó histológicamente. Se reinició la quimioterapia de segunda línea, lográndose una segunda remisión completa que se consolidó con altas dosis de quimioterapia con rescate autólogo con progenitores de sangre periférica. La paciente se mantiene libre de enfermedad 20 meses después del trasplante.
COMENTARIO
El rabdomiosarcoma es el tumor maligno de partes blandas más frecuente en la edad pediátrica. La localización vulvar aislada es muy infrecuente, y representa sólo el 0,9 % de los casos en la serie del Instituto de Patología de las Fuerzas Armadas (AFIP)1 . La mayoría de los rabdomiosarcomas aparecen en la infancia temprana, antes de los 10 años de edad; sin embargo, los casos congénitos son excepcionales2-6 . En un gran estudio multicéntrico de 3.217 pacientes con rabdomiosarcoma, sólo 14 casos fueron congénitos (0,4 %)7 . No hemos encontrado ningún caso en la literatura anglosajona en que se asocien la localización vulvar y la aparición congénita. Desde el punto de vista histológico se pueden distinguir tres tipos principales con pronósticos muy diferentes. El rabdomiosarcoma embrionario es el tipo más frecuente en la edad pediátrica, se localiza habitualmente en cabeza y cuello o aparato genitourinario y es el que tiene mejor pronóstico. El rabdomiosarcoma alveolar ocurre con mayor frecuencia en pacientes mayores, entre 10 y 25 años, y se localiza preferentemente en las extremidades. El tipo pleomórfico es infrecuente y aparece fundamentalmente en mayores de 45 años8 .
El rabdomiosarcoma botrioides es un subtipo del tipo embrionario que representa aproximadamente el 6 % del total de los rabdomiosarcomas 8 . La palabra botrioides deriva del griego botrys (racimo) y eidos (aspecto), ya que se trata de una masa constituida por formaciones polipoides edematosas agrupadas, con una característica apariencia de racimo de uvas. La mayoría de los rabdomiosarcomas botrioides aparecen en órganos huecos con revestimiento mucoso, desde donde pueden extenderse hacia la superficie corporal. El libre crecimiento en cavidades corporales o hacia la superficie explica su apariencia característica. Aunque existen algunos casos descritos, la aparición cutánea primaria es muy rara 4,9-12 . Microscópicamente, el rabdomiosarcoma botrioides muestra una relativa hipocelularidad y una abundancia de estroma mixoide, lo que puede hacernos obviar la naturaleza maligna de la lesión. El rasgo más importante para el diagnóstico es la presencia de la capa de cámbium 1 , denominada así por asemejarse a la capa de células entre la corteza de un tronco y la madera, y que se caracteriza por una condensación subepitelial de células tumorales separadas de un epitelio intacto por una banda de estroma laxo. Las células tumorales varían en apariencia desde células pequeñas primitivas, hasta células con evidente diferenciación rabdomioblástica 1
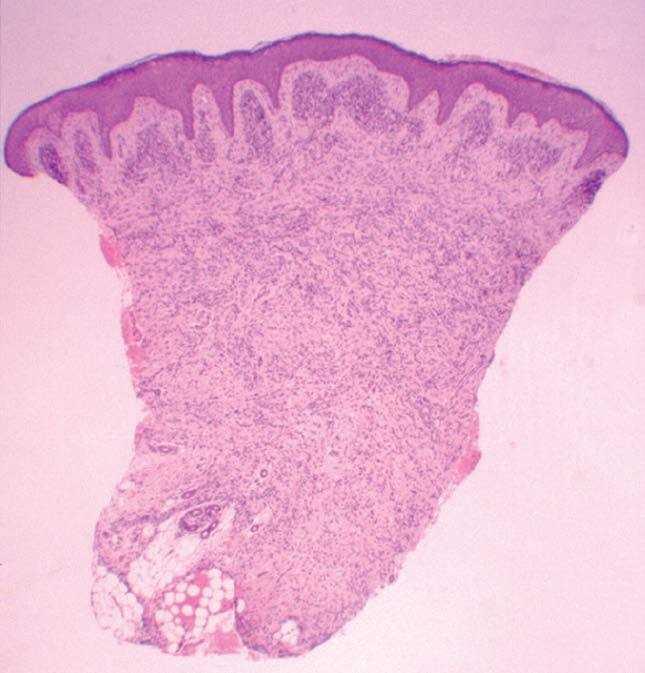
Fig. 2.—La capa de cámbium aparece interrumpida por la presencia de anejos cutáneos. (Hematoxilinaeosina, ×40.)
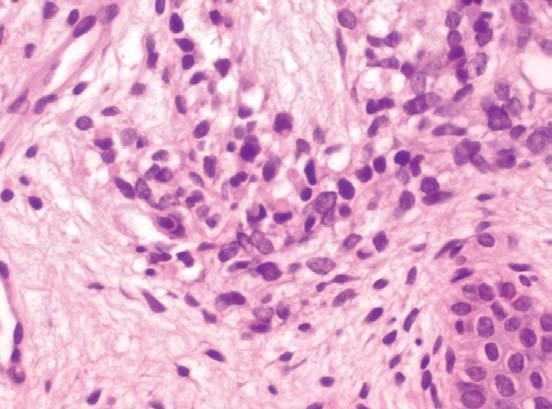
Fig. 3.—Rabdomioblastos redondeados con citoplasma eosinófilo brillante. (Hematoxilinaeosina, ×400.)
La inmunohistoquímica es de gran ayuda en el diagnóstico de este tumor. Dado su origen miogénico, las células neoplásicas son positivas para proteínas musculares como actina, desmina o mioglobina13 . La presencia de estos marcadores se correlaciona con el grado de diferenciación muscular, como ocurre en la miogénesis. Las células más primitivas expresan sólo la vimentina y los marcadores de diferenciación muscular se adquieren progresivamente por los rabdomioblastos en desarrollo. La MyoD1 y la miogenina, factores de transcripción expresados por las células mesequimatosas en su vía de diferenciación hacia músculo esquelético, se utilizan actualmente como anticuerpos estándar para el diagnóstico, ya que son muy sensibles y específicos para el rabdomiosarcoma14 . A diferencia del tipo alveolar, no se ha detectado ninguna alteración genética específica del rabdomiosarcoma embrionario ni de sus variantes15 .
El diagnóstico diferencial con otras lesiones vulva-res en niñas prepúberes incluye una amplia variedad de lesiones, entre las que se encuentran remanentes embrionarios (quiste mesonéfrico, quiste del canal de Nuck, quiste del ducto de Bartholin), hamartomas, hernia inguinal y otros tumores mesenquimatosos benignos (rabdomioma fetal, lipoma, fibroma, hemangioma, etc.) o malignos (tumor del seno endodérmi-co)16 . La característica imagen en racimo de uvas será de gran ayuda para la discriminación entre estas lesiones. El diagnóstico definitivo requiere la confirmación anatomopatológica. La histología aparentemente inocente del rabdomiosarcoma botrioides puede inducir al patólogo a un diagnóstico erróneo. Algunos tumores poco celulares y con abundante matriz mixoide estromal pueden ser malinterpretados como mixomas, lesiones virtualmente inexistentes en niños. Otras entidades benignas que plantean diagnóstico diferencial histológico son el rabdomioma genital y los pólipos fibroepiteliales con células estromales atípicas1 . Si se tienen en cuenta la historia clínica y la ausencia de capa de cámbium, se puede llegar al diagnóstico correcto de la lesión.
El tratamiento del rabdomiosarcoma del tracto genital en niñas se basa en la poliquimioterapia, y no hace falta recurrir a la cirugía radical en la mayoría de los casos. El tratamiento de la enfermedad local es innecesario en aquellas pacientes que muestran una respuesta completa a la quimioterapia, pero si fuera necesario se recomienda el uso de técnicas poco agresivas como cirugía conservadora o radioterapia. Con el empleo de estos esquemas terapéuticos se ha logrando conseguir una alta tasa de curación con una baja incidencia de secuelas funcionales a largo plazo17 .
Correspondencia:
Isabel Colmenero. Servicio de Anatomía Patológica. Hospital Infantil Universitario Niño Jesús. Avda. Menéndez Pelayo, 65. 28009 Madrid. España. icolmenero.hnjs@madrid.salud.org
Recibido el 6 de octubre de 2004. Aceptado el 3 de febrero de 2005.


