


INTRODUCCIÓN
La protoporfiria eritropoyética (PPE) es una enfermedad de origen genético caracterizada clínicamente por fotosensibilidad y analíticamente por un aumento de las protoporfirinas en hematíes, sangre periférica y heces. Los pacientes con PPE tienen un déficit enzimático de la ferroquelatasa implicada en el metabolismo de las porfirinas y provoca el cúmulo de los metabolitos previos a la actuación de la misma, que son los responsables de las manifestaciones clínicas en estos enfermos (fig. 1).

Fig. 1.--Metabolismo del grupo hemo.
Describimos 4 casos de protoporfiria eritropoyética que hemos estudiado en nuestro servicio de Dermatología. Los 4 son niños que tenían en común haber sido interpretados previamente como supuestas alergias al sol y ninguno de ellos había presentado episodios con lesiones clínicas muy evidentes.
DESCRIPCIÓN DE LOS CASOS CLINICOS
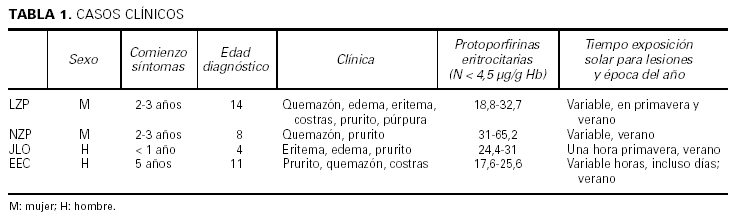
Los casos se resumen en la tabla 1.
Caso 1
La primera paciente era una niña de 14 años sin antecedentes familiares de interés, a excepción de tener una hermana de 8 años que refería síntomas similares (caso 2), hija de padres no consanguíneos. Referían los padres que desde los primeros años de vida presentaba edema, eritema, prurito y calor en cara, manos y pies tras exposición solar en días de verano, muchas veces acompañados de llantos. Los síntomas no ocurrían siempre que se exponía al sol, ni aparecían de la misma manera en cada episodio. Tampoco el intervalo entre la exposición y la clínica era constante. En el momento de la consulta no se objetivó ninguna lesión clínica. Ante la sospecha de que pudiera tratarse de una PPE se realizaron análisis de las protoporfirinas eritrocitarias, que fueron de 22,8 µg/g de hemoglobina (valores normales, hasta 4,5). La analítica realizada a los padres, incluyendo los valores de protoporfirinas eritrocitarias, estaban dentro de la normalidad.
Realizamos un estudio fotobiológico con un simulador solar (Solar Light Company, Filadelfia, Pensilvania), obteniendo una dosis de eritema mínima dentro de los límites normales tanto para UVA como para UVB. Tampoco conseguimos reproducir las lesiones tras 30 minutos de irradiación en su espalda con luz visible de un proyector de diapositivas a 10 cm de distancia.
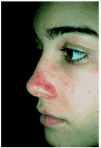
A lo largo de su seguimiento durante 6 años en nuestro servicio la única ocasión en que pudieron apreciarse lesiones objetivables fue en el verano de 2001, cuando la paciente acudió a la consulta por la aparición de lesiones petequiales en dorso nasal tras haber estado de excursión en la montaña y haber descuidado las medidas de fotoprotección (fig. 2). Con la indicación de evitar la exposición al sol, así como el uso de filtros solares y de betacarotenos (40-50 mg/día), permanece prácticamente sin episodios de fotosensibilidad y sin aparición de lesiones residuales. Las analíticas que se le han realizado periódicamente han mostrado una oscilación de las porfirinas entre 18,8 y 32,7 de hemoglobina (normal, hasta 4,5 µg/g de Hb), siendo el resto de las analíticas normales.

Fig. 2.--Lesiones petequiales que presentó una de nuestra pacientes al descuidar las medidas de fotoprotección.
Caso 2
El segundo caso es la hermana de la paciente anterior, de 8 años de edad, quien, al igual que ella, comentaba desde la infancia escozor, quemazón y prurito en región supralabial, manos y pies tras exposiciones solares de intensidad variable. Al igual que en el caso 1, las condiciones en las que aparecían estos síntomas tampoco eran siempre iguales, sino que en ocasiones era inmediatamente tras la exposición y en otras la paciente era capaz de permanecer un tiempo razonable al sol asintomática. El valor de las protoporfirinas eritrocitarias era de 43,7 µg/g de hemoglobina. Los estudios fotobiológicos realizados en las mismas condiciones que en el caso de su hermana fueron normales o negativos.
En los 6 años en los que la hemos seguido en nuestras consultas, la paciente ha permanecido bien controlada con medidas generales de evitación de la exposición directa al sol, uso de filtros solares y betacarotenos. En las analíticas de control los valores de porfirinas eritrocitarias han variado entre 31 y 65,2 µg/g de hemoglobina y el resto de parámetros analíticos han sido siempre normales.
Caso 3
El tercer paciente era un niño de 4 años sin antecedentes personales ni familiares de interés. Los padres negaban casos similares en la familia o consanguinidad entre ellos. Referían, desde los primeros meses de vida, edema, eritema leve y prurito en cara, brazos, manos y pies tras una hora de exposición solar en épocas de primavera y verano. Los episodios se repetían a pesar de la utilización de fotoprotectores y el prurito no mejoraba con la toma de antihistamínicos. Las protoporfirinas eritrocitarias eran de 31 µg/g de hemoglobina y de 43,7 µg/g de heces (normal, hasta 8 µg/g de heces).
Durante los 2 años de seguimiento realizados ha tenido un buen control de los síntomas, evitando la exposición directa al sol, uso de filtros solares y toma de betacarotenos. Los valores de porfirinas han oscilado entre 24,4 y 31 µg/g de Hb, permaneciendo el resto de datos analíticos dentro de los límites normales.
Caso 4
El último paciente era un niño de 11 años que desde que tenía 5 ocasionalmente comentaba tener prurito, escozor y quemazón intensos en manos y región supralabial tras exposición solar en época de verano. A veces ocurría durante los primeros días de verano en los que iba a la playa y otras veces ocurría a los 3 ó 4 días. La madre comentó haber apreciado alguna pequeña lesión costrosa inespecífica en dorso de nariz y orejas. Las protoporfirinas eritrocitarias al diagnóstico eran de 17,6 µg/g de hemoglobina.
Ha realizado tratamiento evitando la exposición al sol, toma de betacarotenos (30 mg/día) y filtros solares con buena respuesta. Los controles analíticos periódicos han sido normales, con variación en los valores de protoporfirinas eritrocitarias entre 17,6 y 25,6 µg/g de hemoglobina.
COMENTARIO
Se cree que el primer caso conocido de PPE fue descrito en 1926 como un hydroa aestivale without haematoporphyrinuria1en un paciente que en 1964 fue diagnosticado de una PPE2. Kosenow y Treibs describieron en 1953 una entidad cutánea que relacionaba una fotosensibilidad inmediata con niveles elevados de porfirinas en sangre3. En 1961, Magnus et al ampliaron la información sobre esta enfermedad y la llamaron protoporfiria eritropoyética4. Describieron el caso de un hombre de 35 años que refería desde los 8 años de edad una historia de intenso escozor en zonas fotoexpuestas inmediatamente tras exponerse al sol.
Se han descrito casos de PPE en todo el mundo, sin diferencias entre razas5ni sexos. No se conoce la incidencia y prevalencia exacta de la PPE en nuestro medio, aunque parece evidente que es una enfermedad poco frecuente. En un estudio extenso realizado en los Países Bajos se recoge una prevalencia de la enfermedad de 1:75.0006, de 1:130.000 en Gran Bretaña7 y de 1:79.000 en Irlanda del Norte8.
El carácter genético de la PPE fue observado en 1963 cuando Haeger-Aronsen describió una familia con tres miembros afectados y dos portadores9. En 1975 se demostró que la actividad de la enzima ferroquelatasa, que es la enzima mitocondrial que cataliza la inserción de los iones férricos en la protoporfirina para formar el grupo hemo7, 10-12, era deficiente en la médula ósea, reticulocitos y fibroblastos cutáneos de estos pacientes13-15. Bloomer et al advirtieron que en cada una de las familias con niños afectados, uno de los progenitores tenía una actividad disminuida de la enzima ferroquelatasa, reforzando así la hipótesis de que éste era el defecto en la PPE16. En un principio, la PPE fue considerada como una enfermedad hereditaria con un patrón autosómico dominante, pero en estudios más extensos se ha visto que el mecanismo de herencia es más complejo6-8,10,12. Así, los pacientes con PPE tienen la actividad de la ferroquelatasa disminuida en un 10%-30% con respecto a los controles13-15 y la transmisión de la enfermedad de padres a hijos ocurre sólo en un 2%-3% de los casos6, 17. Los portadores asintomáticos de la enfermedad presentan un pequeño déficit de la actividad enzimática. Hoy día se cree que para la expresión clínica de la enfermedad es necesaria la conjunción de un alelo mutante y de un alelo con baja expresión18. El gen de la ferroquelatasa se encuentra en el cromosoma 188, 12, habiéndose descrito más de 20 mutaciones distintas19.
Esta deficiencia enzimática lleva a una acumulación de las protoporfirinas que son lipofílicas y se encuentran elevadas en eritrocitos, plasma y heces, siendo característica su ausencia en orina, dada su insolubilidad en soluciones acuosas.
Clínicamente, la PPE se manifiesta como reacciones de fototoxicidad inmediata, a menudo extrema, secundarias a la exposición a la luz solar9, 13. La fotosensibilidad cutánea de la PPE se debe a la interacción de la luz solar con la protoporfirina libre en la piel, que puede absorber fotones de la energía radiante del sol, siendo el espectro responsable el que comprende desde los 400 a los 415 nm10. Generalmente se manifiesta en primavera, verano o principios de otoño en forma de eritema y edema en zonas expuestas a la luz. En ocasiones la fotosensibilidad es leve o moderada y puede no ser obvia clínicamente. Además puede
que no se manifieste en todas las exposiciones e incluso que aparezca a pesar de la fotoprotección. De los casos publicados y de nuestra experiencia parece que existe una relación entre los niveles de protoporfirinas y la intensidad de los síntomas cutáneos. Así, los casos que describimos presentan unos niveles moderados de las mismas que podrían explicar la ausencia de manifestaciones clínicas evidentes.
La PPE generalmente comienza a manifestarse en la infancia, antes de los 5 años, y debe sospecharse por una historia de llanto intenso o dolor en regiones fotoexpuestas tras exposición solar6, 8, 11, 12. Los pacientes refieren una sensación de quemazón, pinchazos, eritema e hinchazón tras una o dos horas de estar expuestos al sol, que pueden recordar clínicamente a lesiones urticariformes, que persisten a lo largo de uno a tres días. Tras exposiciones prolongadas pueden aparecer lesiones purpúricas y/o ampollosas11, 12.
Hay pacientes que refieren que pueden tolerar perfectamente la exposición solar de todo un día sin mayor problema, pero que los primeros minutos de exposición del día siguiente pueden desencadenar los síntomas. Este fenómeno se ha llamado «fenómeno primario», donde la piel se sensibiliza en la primera exposición. Después de exposiciones crónicas muy prolongadas pueden aparecer costras alrededor de los labios o en el dorso de las manos. Como resultado de episodios repetitivos de fotosensibilidad cutánea durante años aparecen cambios cutáneos crónicos de intensidad variable, como engrosamiento céreo y aspecto curtido de la piel (pseudoliquenificación), sobre todo en el dorso de la nariz y las manos, que confieren un aspecto en piel de naranja. También aparecen pequeñas cicatrices en regiones malares y rágades en las comisuras labiales. Las lesiones ampollosas y la hipertricosis no son típicas de la PPE10-12. También es característica de la enfermedad una alta incidencia (12%) de colelitiasis en edades tempranas.
En cuanto a los datos analíticos de los pacientes con PPE, además del incremento de protoporfirinas en plasma, eritrocitos y heces, el 25% de los casos presenta una anemia leve y en un 10%-20% existe un aumento moderado de las enzimas hepáticas y biliares. Esto se debe a la liposolubilidad de la molécula de protoporfirina y su necesidad de excretarse por vía hepática6, 8, 12.
En estudios anatomopatológicos recientes se han analizado las diferencias histológicas entre la piel fotoexpuesta y la no fotoexpuesta en pacientes con PPE. Los hallazgos más característicos son: a) un engrosamiento y una positividad a la tinción con PAS de las paredes de los vasos de la dermis papilar causado por un crecimiento concéntrico de la lámina basal y por un depósito de material granular fino en la zona de la membrana basal de la dermis superficial, y b) la formación de un material amorfo perivascular como consecuencia de la acumulación de distintos componentes séricos. De estos hallazgos parece deducirse que las paredes de los vasos de la dermis superficial son las principales estructuras en resultar dañadas durante los episodios de exposición solar10.
El daño tisular agudo repetido y el proceso de reparación en la zona de la membrana basal provocan un engrosamiento de las paredes vasculares y se pueden observar estas imágenes en todos los tipos de porfirias cutáneas. Los depósitos perivasculares amorfos son un fenómeno secundario e irreversible que resulta de la acumulación de distintos componentes séricos durante la fase aguda; estos cambios son permanentes y no varían durante las fases asintomáticas. Todos estos hallazgos enumerados no se encontraron en las biopsias de piel no fotoexpuesta de los pacientes con PPE, de lo que se puede deducir que la interacción de las protoporfirinas y la radiación solar es necesaria para que se den estos cambios histológicos, y no son suficientes los altos valores de protoporfirina para dar lugar a estas alteraciones.
El diagnóstico de la PPE se establece con los datos clínicos característicos y se confirma con los hallazgos analíticos que demuestran el incremento de las protoporfirinas en sangre y en heces. El diagnóstico diferencial más importante debe establecerse con la urticaria solar, de manera que en todos los pacientes con urticaria solar debe solicitarse análisis de porfirinas para establecer el diagnóstico definitivo.
La complicación más seria en los pacientes con PPE es la del fallo hepático fulminante. Se produce por acumulación de protoporfirinas en el hígado y ocurre en menos de un 5% de los pacientes con PPE20-23. Consiste en un fallo rápidamente progresivo con marcadas elevaciones en la concentración de protoporfirinas en plasma, aumento de coproporfirinas urinarias y unas protoporfirinas fecales descendidas.
Según Bloomer, la concentración de protoporfirinas podría alcanzar unos niveles que sobrepasaran la capacidad del hígado para solubilizarla, causando un exceso de protoporfirinas libres que podrían agregarse y formar depósitos sólidos en los hepatocitos y en los pequeños canalículos biliares24, 25. La obstrucción de los canalículos biliares por los cristales de porfirina establecería un círculo vicioso en la patogenia de la enfermedad.
Dada la disminución del aclaramiento de protoporfirinas por el hígado, el patrón de excreción de porfirinas cambia en el fallo hepático, aumentando las eritrocitarias y disminuyendo el nivel de protoporfirinas fecales. Las porfirinas urinarias aumentan, sobre todo las coproporfirinas. En la mayoría de los casos el primer signo de fallo hepático es la aparición de ictericia, y muchos pacientes refieren un dolor abdominal que irradia a la espalda26. Dado que la colestasis suele ser el signo de este fallo orgánico, Doss y Frank consideran como parámetros de fallo hepático inminente el aumento de porfirinas en sangre, el descenso en heces y el aumento de coproporfirinuria a expensas del isómero I27. Bloomer recomienda realizar biopsia hepática en todo paciente cuyas tasas de protoporfirina superen los 1.500 µg/dl en hematíes o los 50 µg/dl en plasma24.
El gran problema de esta complicación es que se presenta súbita e insidiosamente y los factores que influyen en este proceso son desconocidos.
No hay un método para predecir el riesgo de enfermedad hepática grave en pacientes con PPE, aunque en varios artículos consultados se recomiendan análisis de enzimas hepáticas de control cada 6 meses.
El trasplante hepático parece ser una buena alternativa para el tratamiento de esta complicación, pero existe una experiencia limitada. Los pacientes pueden sufrir complicaciones postoperatorias serias y el pronóstico de la enfermedad metabólica a largo plazo no se conoce bien10, 11, 19, 28.
El primer caso publicado de trasplante hepático en un paciente con PPE en España se realizó en 199519. Hasta 1999, 31 pacientes con PPE habían recibido un trasplante hepático, con una alta tasa de supervivencia. Ya que la médula ósea es considerada la causante de esta enfermedad por la excesiva producción de protoporfirinas, la opción lógica de tratamiento sería la sustitución completa de la población endógena celular por un sistema de células madre eritropoyéticas normales. El trasplante combinado de hígado y médula ósea sería el tratamiento teórico de esta enfermedad, pero al no haber una forma de predecir el fallo hepático en un paciente con PPE, y dados los riesgos que conlleva un trasplante de médula ósea, se ha desestimado la realización de éste de manera preventiva29. No hay experiencia sobre el trasplante de médula ósea en pacientes con PPE, pero se ha realizado en ratas, con una corrección completa a largo plazo de la fotosensibilidad30.
Tratamiento
La faceta más importante del tratamiento de la PPE consiste en evitar la exposición directa a la radiación solar. Los filtros solares no suelen ser eficaces en la prevención de las lesiones por su limitada capacidad de filtro del espectro visible responsable de la fotosensibilidad de estos pacientes. También se utilizan frecuentemente los betacarotenos por su capacidad de neutralizar los radicales libres, pero su mecanismo de acción no se conoce bien por el momento. A pesar de su amplio uso, no hay datos concluyentes acerca de su eficacia. La dosis media es de 30 a 180 mg/día, para mantener los niveles séricos de carotenos entre 600 y 800 mg/dl31.
Aunque el trasplante hepático es el tratamiento de elección en los casos de PPE asociada a fallo hepático, se han propuesto varias líneas de tratamiento, sin mucho éxito, para intentar evitar la acumulación de protoporfirinas en el hígado en dicha situación, como disminuir la producción excesiva de protoporfirinas (hierro oral, administración intravenosa de hematina, transfusión de sangre o dieta rica en carbohidratos)32, 33, facilitar el transporte hepático y secreción de protoporfirinas en la bilis mediante administración oral de ácidos biliares34e interrumpir la circulación enterohepática de la protoporfirina con colestiramina o carbón activado35.
También se ha descrito en algunos artículos aislados la mejoría de estos pacientes con cisteína36, 37, vitamina E y C (como antioxidantes) y el recambio de células eritrocitarias con células autólogas lavadas (para eliminar protoporfirinas)10, 11, 28, 38.
Últimamente se postula la posible eficacia de psoraleno (PUVA) y luz ultravioleta (UVB) de banda estrecha para inducir una pigmentación cutánea que favoreciera la fotoprotección natural del espectro responsable de la fotosensibilidad en la PPE7, 10-12, 38, 39. Otra de las opciones sería la eliminación de la fuente de protoporfirinas a través de un trasplante de médula ósea, pero esto no se ha llegado a realizar nunca debido al riesgo de enfermedad de injerto contra huésped que pudiera atacar al hígado10, 40.
Hasta el momento no se han visto empeoramientos de los niveles de protoporfirinas secundarios a fármacos, aunque en un artículo se mencionaban la azatioprina, ciclosporina A y el tacrólimus como posibles causantes de empeoramiento de estos niveles en un paciente trasplantado de hígado22.


