


INTRODUCCION
La porfiria hepatoeritrocitaria es una entidad nosológica harto infrecuente, descrita inicialmente por Gunther en 19671 y en 1969 por Piñol Aguadé2, cuya evolución a lo largo de los años es todavía poco conocida. Presentamos la evolución durante 9 años de una niña diagnosticada durante primera infancia y se realiza una revisión de casos en la literatura.
DESCRIPCION DEL CASO
Una niña de 10 años de edad, segunda de cuatro hermanos, nacida tras embarazo y partos normales, presentó al año de vida ampollas y erosiones en el dorso de las manos y la cara, que empeoraban con la fotoexposición y curaban dejando cicatriz e hiperpigmentación (fig. 1). Al inicio, se acompañó de varios episodios autorresolutivos de orinas rojas. Los estudios analíticos practicados revelaron un leve déficit de hierro sérico (46 mg/dl; normal, 50-150 mg/dl), con niveles muy elevados de porfirinas en orina y heces (porfirinas totales; 9.935 μg/24 h; uroporfirinas: 7.282 μg/24 h; coproporfirinas: 2.653 μg/24 h). En eritrocitos se evidenció un descenso marcado (3,88 U) (menor del 10 % de los valores normales) de la actividad de la urogendescarboxilasa.
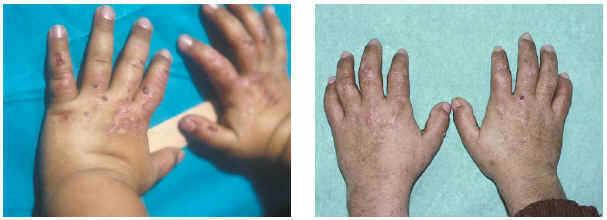
Fig. 1.--Lesiones ampollosas y erosivas.
Una vez realizado el diagnóstico de porfiria hepatoeritrocitaria, se inició tratamiento con dosis bajas de cloroquina (250 mg/2 veces por semana) y fotoprotección, tanto con medidas físicas como con filtros solares. Pese a esto, las lesiones continuaron apareciendo, aunque en menor número, con formación de ampollas y desarrollo de cicatrices, sobre todo en dorso de manos, dedos y cara, produciéndose deformidad de predominio nasal. Progresivamente la paciente fue desarrollando hipertricosis malar y frontal muy evidentes (fig. 2). Analíticamente, se produjo una mejoría parcial de los valores de uro y coproporfirinas (octubre de 2002: porfirinas totales, 6.361 μg/24 h; uroporfirinas: 4.874 μg/24 h; coproporfirinas: 1.487 μg/24 h), que no llegaron a normalizarse en ningún momento; la anemia inicial se corrigió espontáneamente y no presenta alteraciones del perfil hepático.
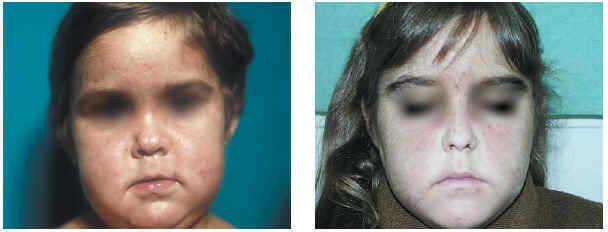
Fig. 2.--Hipertricosis malar.
La paciente lleva una vida escolar normal para su edad y se relaciona con otros niños de modo adecuado, excepto en lo que se refiere a las actividades de la calle, que son limitadas para evitar la fotoexposición. Tiene otros tres hermanos, de once, ocho y un año de edad, que hasta el momento no han presentado síntomas.
DISCUSION
Los primeros casos descritos de la porfiria que posteriormente se denominaría hepatoeritrocitaria corresponden a Gunther (1967)1 y a Piñol Aguadé2 (1969). En 1981, Elder et al3 descubrieron la actividad deficiente de la urogendescarboxilasa en los pacientes con porfiria hepatoeritrocitaria, la misma enzima deficitaria que en la porfiria cutánea tardía, pero con valores del 10 % y del 30-50 % de los normales, respectivamente4. En la actualidad se postula una herencia autosómica recesiva polimutacional en el brazo corto del cromosoma 1, con más de nueve deleciones/mutaciones descritas para esta entidad5. La sintomatología es precoz, con un inicio en la primera infancia con marcada fotosensibilidad y formación de ampollas y cicatrices, así como hipertricosis, y ocasionalmente presenta asociación de eritrodoncia, anemia hemolítica, orinas rojas y hepatoesplenomegalia; analíticamente se caracteriza por un aumento de uro y coproporfirinas6.
Son muy escasos los pacientes de los que se haya publicado su evolución en el tiempo. Czarnecki7 publicó en 1980 los resultados del seguimiento de una serie de 7 pacientes, 5 mujeres y 2 varones, de edades comprendidas entre los cuatro y los 60 años de edad; en todos encontró un inicio de la enfermedad durante el primer año de vida, con formación de eritema, ampollas y cicatrices tras fotoexposición, que en ocasiones llegaban a deformar los rasgos faciales (nariz, orejas) y las manos (flexura parcial, acortamiento digital), de modo similar a la esclerodermia. También se acompañaban de hipertricosis facial infantil y la mayoría de los pacientes, durante el verano, presentaban episodios de orinas rojas. Con el tiempo y el predominio cicatrizal, algunos de estos pacientes presentaban una mejoría espontánea de la fotosensibilidad, que no se acompañaba de una mejora analítica. Asimismo se señalaba la posibilidad de complicaciones sistémicas como hepatopatía y alteración del perfil hepático y anemia (moderada y normocrómica) en probable relación con un defecto intrínseco de la eritropoyesis. En cuanto al tratamiento, ni siquiera las medidas fotoprotectoras parecen ser capaces de controlar la enfermedad8. Nuestra paciente, durante estos 9 años de seguimiento, ha evolucionado de forma similar con formación de cicatrices en áreas fotoexpuestas, hipertricosis facial, afilamiento digital y leve anemia con valores normales de hierro sérico, pero sin mejora de la fotosensibilidad ni de los valores de porfirinas.


