



Sr. Director:
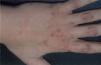
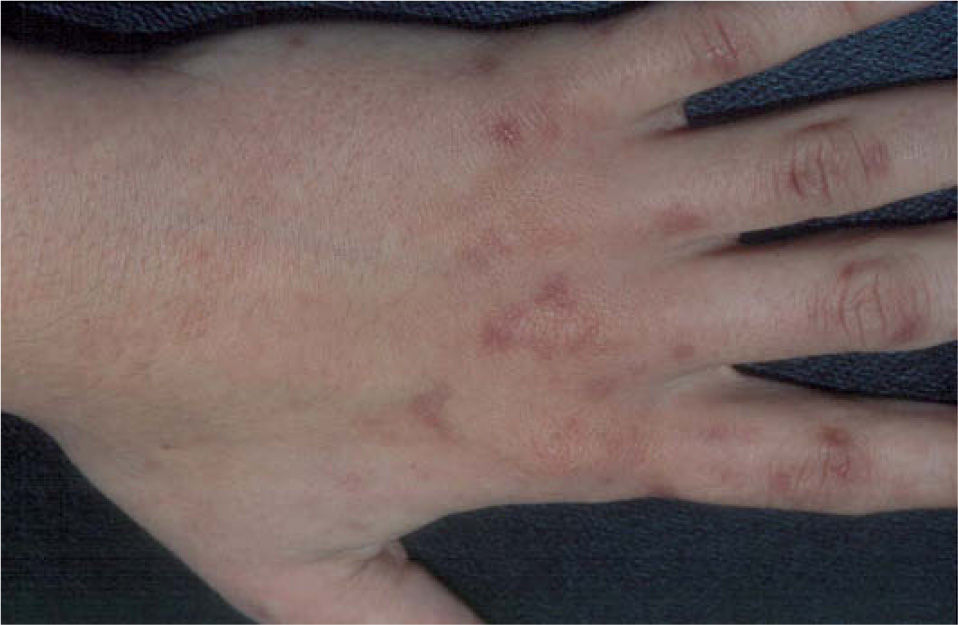
Presentamos el caso de una mujer de 31 años sin antecedentes personales de interés, que consultó por lesiones en el dorso de las manos, que relacionaba con pequeños traumatismos, y que se acentuaban en la época estival, dejando áreas hiperpigmentadas al resolverse (fig. 1). La paciente tenía un fototipo III y presentaba una discreta hipertricosis en las regiones cigomáticas. Con la sospecha de una dermatosis fotosensible, se pidió una batería de pruebas que incluían determinación de enzimas hepáticas, hierro sérico, niveles de transferrina y ferritina, porfirinas en sangre y orina y la realización de fototest.
Los niveles de porfirinas en la orina de 24 horas estaban elevados: 2.104μg/l (valores normales: inferiores a 200μg/l), de los cuales el 85 % eran uroporfirinas.
En plasma los valores de porfirinas de 25μg/l estaban por encima del rango normal (menor de 10μg/l). Las cifras de hierro sérico: 150μg/dl (valores normales: 92-155μg/dl), transferrina: 310mg/dl (valores normales: 205-365mg/dl) y ferritina: 175μg/ml (valores normales: 13-160μg/ml) eran elevadas o en el límite alto de la normalidad. La ecografía hepática no demostró alteraciones.
El diagnóstico fue de porfiria cutánea tarda (PCT) teniendo en cuenta los síntomas y signos clínicos, y los parámetros analíticos. La paciente no refería antecedentes familiares de lesiones cutáneas ni de hepatopatías. Los anticonceptivos orales (ACO) que había tomado hasta 6 meses antes de acudir a la consulta se consideraron como posible agente desencadenante de la PCT, sin embargo, tras la suspensión las lesiones continuaron apareciendo. No parecía que existieran otros factores desencadenantes ya que no bebía alcohol habitualmente y las serologías de virus hepatotropos eran negativas.
El estudio genético para las mutaciones más frecuentes de hemocromatosis (C282Y, H63D) mostró heterocigosidad para C282Y, lo que podría explicar las alteraciones discretas que se observaron en el metabolismo del hierro y que podrían haber contribuido, junto a la toma de ACO, al desarrollo de la PCT. Ante el deseo de la paciente de tener descendencia se realizó el estudio genético para las mismas mutaciones de hemocromatosis en el marido, que demostró heterocigosidad para la mutación H63D.
El tratamiento consistió en flebotomías con una periodicidad quincenal, con remisión de las lesiones cutáneas y normalización de los valores de porfirinas en el plasma y la orina.
No nos fue posible realizar la determinación de la actividad enzimática de la uroporfirinógeno decarboxilasa (URO-D) eritrocitaria por pérdida en el seguimiento de la paciente.
La PCT se debe a un déficit o inactivación de la enzima URO-D, que da lugar a la acumulación de metabolitos fotosensibles que son excretados en la orina y las heces1.
Se distinguen 3 tipos: I, II y III2. El tipo I o esporádico es el más frecuente, en él la inactivación solo se produce en el hígado y no existe historia familiar previa. El tipo II o familiar se caracteriza porque la inactivación o déficit de la enzima URO-D se produce en todos los tejidos. El tipo III se caracteriza porque la inactivación se produce en el hígado pero se acompaña de historia familiar previa.
Suele iniciarse en la edad adulta, y se conocen diversos factores desencadenantes: hepatitis víricas, alcohol, ACO y terapia hormonal sustitutiva (THS), hidrocarburos policlorados, hemodiálisis y situaciones que dan lugar a sobrecarga férrica como la hemocromatosis3,4. En ocasiones son varios los factores implicados2,4. El agente desencadenante más frecuente varía según la edad, el sexo y el área geográfica. En los hombres los más comunes son el abuso de alcohol y las hepatitis víricas crónicas, mientras que, en las mujeres, es la terapia hormonal, constituyendo en un porcentaje importante el único factor implicado. La exposición a hidrocarburos se observó en países subdesarrollados, mientras que la infección por el virus de la hepatitis C es más frecuente en países mediterráneos y latinoamericanos2.
La hemocromatosis es una enfermedad genética con una herencia autosómica recesiva y una prevalencia de 1/2005. Se caracteriza por un aumento de la absorción intestinal de hierro y la acumulación del mismo en los tejidos (hígado, páncreas, miocardio, piel, articulaciones), donde producirá manifestaciones clínicas. La sospecha de hemocromatosis en mujeres premenopáusicas se realiza ante una cifra de saturación de la transferrina superior al 50 % o con valores de ferritina superiores a 200μg/l6. Aunque el 80 % de los pacientes con PCT tiene siderosis hepática y el 60 % tiene hipersideremia, menos del 20 % cumple criterios de hemocromatosis7.
Recientemente se han identificado varias mutaciones relacionadas con dicha enfermedad8. Las más frecuentes están en el gen HFE situado en el brazo corto del cromosoma 6, y son la C282Y y la H63D. En la primera se sustituye una tirosina por una cisteína en el aminoácido 282, mientras que en la otra se cambia la histidina por ácido aspártico en la posición 638. El 80 % de los enfermos es homocigoto para C282Y, mientras que el 20 % es doble heterocigoto para las dos mutaciones anteriores.
La prevalencia de estas mutaciones varía entre las poblaciones. La primera predomina en países nórdicos y centroeuropeos, y en España, en las poblaciones de origen celta (Galicia y Asturias)9, mientras que la segunda es frecuente en el área mediterránea. En España, la prevalencia de C282Y es de 1,7 %, mientras que la de H63D es de 20 %10. Sin embargo, el diagnóstico clínico de hemocromatosis es mucho menos común, lo que hace suponer que su penetrancia es baja y que el diagnóstico clínico puede ser tardío.
Estas mutaciones parecen más frecuentes en pacientes con PCT debido a que los homocigotos, heterocigotos y dobles heterocigotos para estas mutaciones tienen más tendencia a la sobrecarga de hierro que actúa como desencadenante de la PCT, ya sea de forma única o concomitante. La mutación H63D en heterocigosis tendría poca importancia en la sobrecarga férrica y por tanto en el desarrollo de la PCT, sin embargo su papel se incrementa en la doble heterocigosis4.
Varios estudios con series de pacientes han demostrado la asociación anterior. Así, en la mayor parte de Europa, incluida España, EE.UU. y Sudamérica la mutación más frecuente es la C282Y3,4,10–16, mientras que en Italia es la mutación H63D17 (tabla 1).
Estudios que incluyen series de pacientes sanos y con porfiria cutánea tarda (PCT) donde se observa que las mutaciones C282Y y H63D son más frecuentes en los pacientes afectos de PCT
| Estudio | Mutación | PCT | Controles | País |
| Roberts (1997) | C282Y | 44 % | 11 % | Reino Unido |
| Santos (1997) | H63D | 23 % | 4 % | Holanda |
| Sanpietro (1998) | H63D | 29 % | 13 % | Italia |
| Martinelli (2000) | C282Y | 17 % | 4 % | Brasil |
| Bulaj (2000) | C282Y | 34 % | 13 % | EE.UU. |
| H63D | 22 % | 20 % | ||
| Chiaverini (2003) | C282Y | 18 % | 1 % | Francia |
| H63D | 54 % | 37 % | ||
| Frank 2006 | C282Y | 15 % | 5 % | Alemania |
| H63D | 35 % | 29 % |
El tratamiento de elección para la PCT son las flebotomías con una periodicidad quincenal, extrayendo 200-500ml según la tolerancia. La respuesta clínica se obtiene a los 2-3 meses y la normalización analítica a los 12 meses. Se considera que cifras de hierro sérico menores de 25μg/l son suficientes para el control de la enfermedad18. Otras opciones terapéuticas aceptadas son los antipalúdicos cloroquina e hidroxicloroquina que, asociados a las sangrías, consiguen respuestas más rápidas3,17. La respuesta a los antipalúdicos es menor si la sobrecarga férrica es importante, lo que suele corresponder a pacientes homocigotos para C282Y, por tanto, en ellos puede ser necesario asociar las dos opciones previas3,18.
Es fundamental corregir o minimizar los factores desencadenantes como la terapia hormonal, el alcohol, la insuficiencia renal y las hepatitis víricas.
Con este artículo se pretende llamar la atención sobre la conveniencia de realizar el despistaje genético de hemocromatosis en aquellos pacientes con PCT en los que no se encuentre un factor desencadenante claro, ya que es una prueba disponible en nuestra sanidad que puede ser útil para diagnosticar estados de hemocromatosis latentes. Además, al ser mutaciones relativamente frecuentes (hasta un 20 % de la población puede ser portadora) se podría hacer un estudio y consejo genético en las parejas en las que uno de los miembros presente PCT y algunas de las mutaciones ya descritas11.
En este caso los dos miembros de la pareja eran portadores para las dos mutaciones más frecuentes de hemocromatosis, por lo que la posibilidad de que su descendencia sea doble heterocigota es de un 25 %, si bien es cierto que la penetrancia de esta enfermedad es baja y por tanto la posibilidad de padecer hemocromatosis será menor.
En esta paciente no se pudo establecer con seguridad el subtipo de PCT ya que, aunque no existía historia familiar previa de fotodermatosis ni de enfermedad hepática, no fue posible realizar la determinación de URO-D eritrocitaria ni tampoco el estudio genético de la URO-D, por lo tanto no se puede excluir la PCT de tipo II. Aunque esto no es importante para el manejo terapéutico, sí que es útil de cara a establecer un consejo genético, sobre todo en mujeres jóvenes afectadas, en las que no existan otros factores desencadenantes19.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.


