


INTRODUCCIÓN
El mieloma múltiple es una neoplasia de células plasmáticas maduras, responsable de un 10% de los procesos oncohematológicos y que produce afectación cutánea específica (plasmocitoma) en un 5-10% de los casos. Excepcionalmente es la primera manifestación de la enfermedad (1-3).
DESCRIPCION DEL CASO
Paciente de 55 años que ingresó desde urgencias en el Servicio de Cirugía de Tórax por molestias y drenaje de líquido seroso en una masa torácica preesternal.
El paciente, previamente sano, presentó 2 meses antes una tumoración en la pared anterior del tórax, asintomática durante todo este período y que fue aumentado progresivamente en volumen hasta alcanzar el tamaño actual.
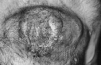
En la exploración física el enfermo presentaba un buen estado general, observándose en pared anterior del tórax una tumoración de aproximadamente 15 cm de diámetro mayor que se elevaba unos 3-4 cm sobre la superficie cutánea, de coloración violácea, superficie irregular erosionada, consistencia dura con áreas de necrosis en su interior y gran infiltración de la base (Fig. 1). El resto de la exploración física estuvo dentro de la normalidad.
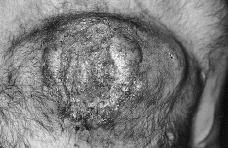
FIG.1.--Lesión tumoral en tórax correspondiente a un plasmocitoma cutáneo secundario a mieloma múltiple.
Entre las pruebas complementarias realizadas destacamos los siguientes datos. En sangre periférica se encontraron una leucocitosis de 5.220 (segmentados, 91%; linfos, 2%; monos, 7%), hematíes, 3.320.000; hemoglobina, 10,1 g%; hematócrito, 29,9; VCM, 89; CHM, 30,6; CHCM, 34; plaquetas, 238.000. En la bioquímica sanguínea: creatinina, 1,2; calcio, 9,2; úrico, 7,2; LDH, 209; proteínas totales, 7,5 g%; albúmina, 3,5 g%; resto de parámetros dentro de la normalidad. Orina elemental normal. Cuantificación de inmunoglobulinas: IgG, 2.060 mg%; IgA, 27 mg%; IgM, 26 mg%. Inmunoelectroforesis en suero: IgG muy aumentada y de morfología monoclonal, IgA muy disminuida, IgM muy disminuida, cadenas ligeras lambda muy aumentadas y de morfología monoclonal frente a cadenas kappa. Beta-2 microglobulina, 5,9 mg/l. Inmunoelectroforesis en orina: proteinuria de 0,05 g%. No eliminaba proteína de Bence-Jones.
Seríe ósea radiológica: en placa de tórax aparecía una masa de partes blandas que destruía parte del esternón y se extendía a mediastino. Se observaban imágenes líticas en tercera, séptima y novena costillas del lado derecho, así como en arco costal de la octava izquierda. En pelvis una imagen lítica en rama isquiopubiana del lado derecho. Cráneo normal.
La TAC torácica mostraba una gran masa sólida en pared anterior que destruía parcialmente el cuerpo del esternón y ocupaba el compartimento mediastínico anterior, rechazando estructuras vasculares y con engrosamiento de la pleura mediastínica anterior. No se encontraron adenopatías.
La ecografía abdominal mostraba en lóbulo hepático derecho varias calcificaciones puntiformes, un riñón izquierdo con pequeño quiste de 1 cm y el resto dentro de la normalidad.
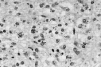
Tras punción-aspiración orientativa se realizó biopsia de la masa torácica, observándose una abundante celularidad atípica dispuesta en sábana, principalmente en dermis profunda e hipodermis, de hábito plasmocelular, constituidas por células de núcleos generalmente excéntricos, redondeados, grandes, con uno o dos nucléolos prominentes y mostrando un citoplasma finamente granular de frecuente disposición en babero y aisladamente células plasmáticas maduras entre la celularidad blástica (Fig. 2). El estudio inmunohistoquímico mostró una infiltración lambda monoclonal sugestiva de mieloma.
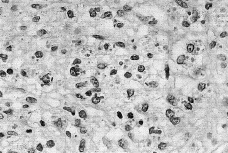
FIG. 2.--Infiltrado monomorfo difuso de células plasmáticas de specto blástico.
En la médula ósea obtenida por punción de cresta ilíaca posterior izquierda se encontró una celularidad normal, megacariocitos normales, pero con un 18-23% de la celularidad constituida por células plasmáticas atípicas, con núcleo de cromatina inmadura y nucléolo evidente, clasificables como plasmoblastos. La serie mieloide, bien representada (52-67%) en todos los estadios evolutivos y la línea eritroide (1-7%) con rasgos de diseritropoyesis. Había discreta linfocitosis en algunas zonas. La tinción de Perls mostró muy importantes depósitos de hierro, sideroblastos con múltiples gránulos (42%) y sideroblastos en anillo (4%).
El diagnóstico a la luz de todos estos datos fue el mieloma múltiple IgG lambda con lesión específica cutánea de gran tamaño.
Se inició tratamiento con VAMP (vincristina, 1,6 mg; adriamicina, 48 mg, y metil prednisolona, 1,5 g por 5 días), con respuesta pobre y rápido recrecimiento, por lo que se instauraron tres ciclos de CHOP, con los que se obtuvo respuesta completa, evidenciándose solamente en la zona de la masa un área eritematosa de unos 10 cm de diámetro sin sobreelevación ni infiltración.
A los 2 meses se realizó trasplante autólogo de médula ósea. Tras una respuesta inicial con regresión total de la masa y médula ósea libre se produjo un recrecimiento en forma de nódulo subcutáneo de 2,2 cm de diámetro en la misma localización de la masa inicial que había demostrado sensibilidad inicial a CHOP. Fue radiada, con lo que desapareció por completo. A las 4 semanas recreció una nueva masa localizada en el cuadrante superior izquierdo de la masa original, que llegó a alcanzar los 8 cm, con una morfología nodular, superficie irregular, indurada y algo dolorosa. La médula ósea persistía libre de células neoplásicas. A los 7 meses se realizó nuevo trasplante autólogo de médula ósea, con lo que se obtuvo una mejoría evidente, con desaparición total de las masas descritas.
Tras un intervalo libre de unos meses vuelven a recrecer las masas, iniciándose tratamientos con M-2, dexametasona intermitente, VMCP, VCAP. Todas se mostraron inútiles, apareciendo recrecimiento tumoral interciclo. El paciente falleció a los 18 meses del diagnóstico por progresión de la masa tumoral.
DISCUSION
En 1955 fueron descritas por Bluefarb (4), de forma sistemática, las manifestaciones cutáneas del mieloma múltiple. Las dividió en lesiones cutáneas específicas e inespecíficas. Las primeras son los plasmocitomas: lesiones poco frecuentes que aparecen entre un 5-10% de todos los mielomas múltiples, siendo la forma más frecuente de presentación las debidas a diseminación directa por un foco óseo subyacente de mieloma múltiple, como en nuestro caso, y menos frecuentemente por metástasis a distancia del tumor (5-13).
Los plasmocitomas secundarios a mieloma múltiple suelen presentarse como lesiones solitarias o múltiples de aspecto cupuliforme, color angiomatoso, de 1 a 5 cm y localizadas habitualmente en cabeza, extremidades superiores y tronco (12, 14-16).
En la histología, aparte del patrón en sábana, difuso, infiltrativo encontrado en nuestro paciente, puede aparecer un patrón macronodular profundo, constituido por masas cohesivas de células plasmáticas (17). Con respecto a la celularidad de la que se componen estos tumores, ésta es a veces muy difícil de diferenciar, por la gran inmadurez de sus células, el aspecto blástico, todo lo cual puede llevarnos a confusión con otros tumores como los linfomas o los melanomas; en todo caso habitualmente se encuentran claves para el reconocimiento de la población plasmocelular, como la morfología en babero de las células, el núcleo excéntrico, la cromatina dispuesta en rueda de carro, etc. (18). En otro orden de cosas se nos plantea la difícil diferenciación entre una proliferación tumoral de las células plasmáticas y una proliferación simplemente reactiva de esas células. En este último caso la inmunohistoquímica con la demostración de restricción de cadenas ligeras nos ayudarán en el diagnóstico (17, 18).
La presencia de un plasmocitoma cutáneo en el seno de un mieloma múltiple implica mal pronóstico. En más del 50% de estos pacientes las lesiones se producen en estadios tardíos de la enfermedad cuando la carga tumoral es muy elevada (5, 7, 19, 20). Alberts y Lynchs relacionaron la aparición de lesiones cutáneas específicas y la carga tumoral en hueso, de manera que cuando esta última alcanza los 2-3 kg la probabilidad de lesiones cutáneas es muy alta (21).
El caso que presentamos reúne una serie de características atípicas con respecto a otros ya documentados. Se trata de un plasmocitoma secundario a mieloma múltiple de «gran tamaño», «gigante» (1, 2), primera manifestación de la enfermedad subyacente del paciente, con alta tendencia a la recidiva y gran resistencia a la terapéutica quimioterápica convencional, características que proporcionan un pésimo pronóstico a corto plazo.


