


HISTORIA CLINICA
Un hombre de 59 años de edad, con antecedentes personales de psoriasis y sarcoidosis pulmonar de larga evolución, tratada durante muchos años con elevadas dosis de corticoides orales (hasta 210 mg/día), consultó por una lesión asintomática, de 3 meses de evolución y rápido crecimiento, en región frontotemporal izquierda.
EXPLORACION FISICA
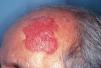
En la exploración física se objetivó una placa de gran tamaño (5 x 6 cm), apenas sobreelevada, de coloración uniforme eritematosa violácea (fig. 1). La lesión presentaba una superficie lisa y brillante y tenía bordes irregulares, más o menos bien delimitados. La línea de implantación del pelo no alcanzaba los bordes clínicos de la lesión, dejándola totalmente expuesta. No se hallaron adenopatías ni visceromegalias.
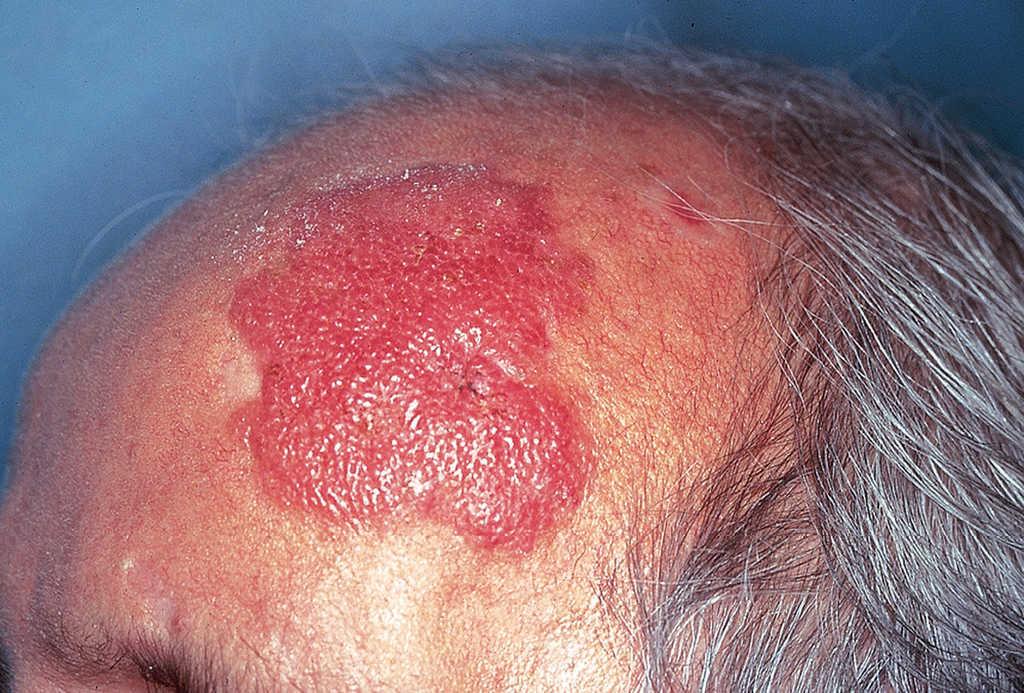
Fig. 1.--Placa eritematosa violácea de gran tamaño en zona frontotemporal izquierda.
EXPLORACIONES COMPLEMENTARIAS
Se realizó una biopsia de la placa frontal (fig. 2). Los análisis, incluyendo hemograma, bioquímica, coagulación y serología para el virus de la inmunodeficiencia humana (VIH) fueron normales o negativos. Se realizó una radiografía de tórax, una TC cervicotoracoabdominal y una RM cerebral, que no revelaron hallazgos patológicos.
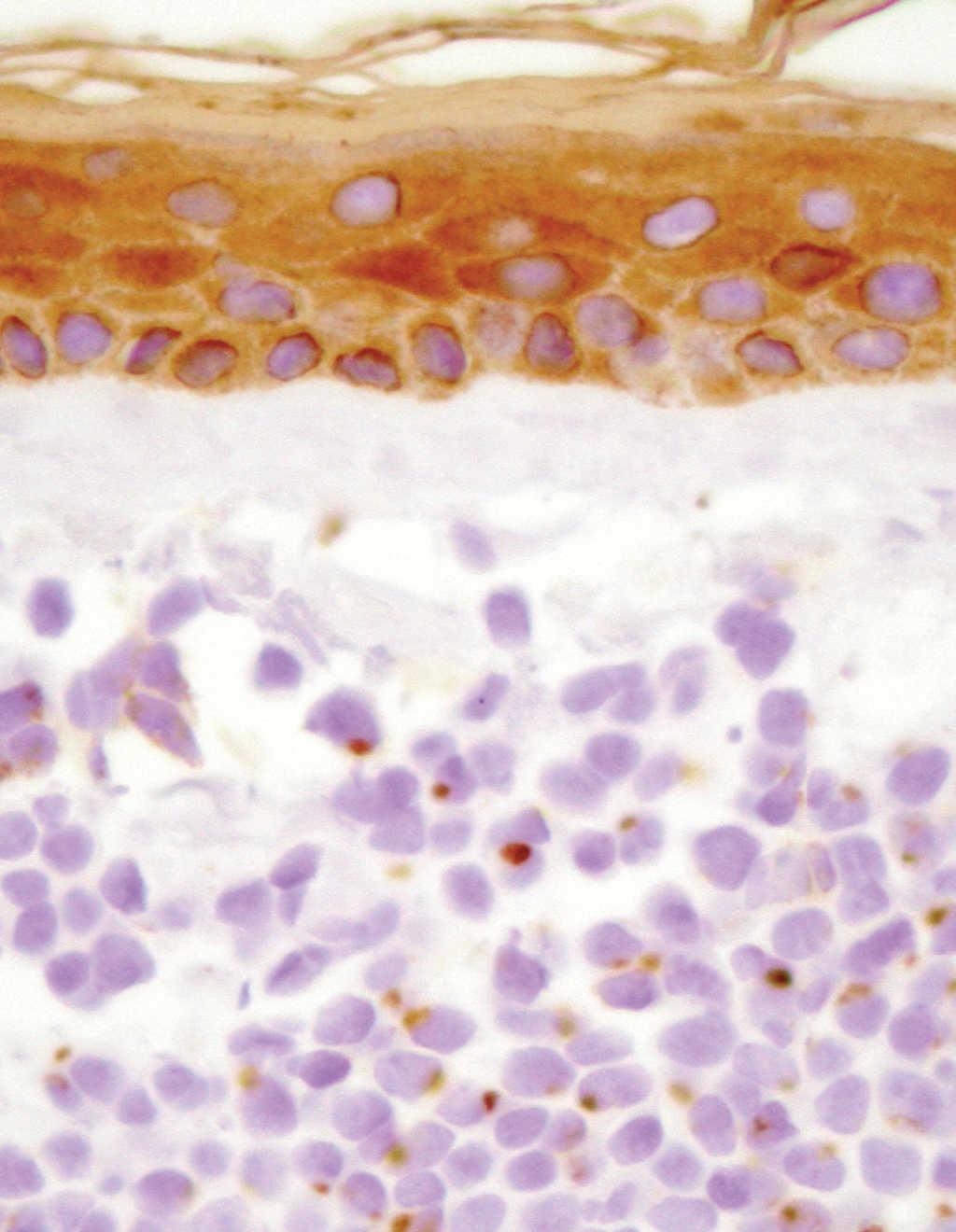
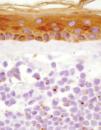
Fig. 2.--Detalle de la tumoración en dermis superficial, con una evidente positividad paranuclear en gotas con citoqueratinas (AE1-AE2).
DIAGNOSTICO
Tumor de células de Merkel.
HISTOPATOLOGIA
El estudio histopatológico mostró infiltración tumoral de la dermis por células redondeadas de tamaño pequeño e intermedio, con escaso citoplasma, y que se disponían de forma compacta en nidos. El estudio inmunohistoquímico demostró positividad para citoqueratinas (AE1-AE2) y neurofilamentos, con un patrón paranuclear en gotas (fig. 2). También se obtuvo positividad con las tinciones para sinaptofisina y cromogranina A.
EVOLUCION Y TRATAMIENTO
Se procedió a su exéresis quirúrgica con un margen lateral de 2 cm y hasta periostio en profundidad, seguido de radioterapia local con una dosis total de 50 Gy y fraccionamiento de 2 Gy. Aproximadamente 6 meses después de finalizar el tratamiento radioterápico, se detectó diseminación metastásica hacia la cadena ganglionar parotídea izquierda, que requirió linfadenectomía regional y régimen adyuvante de radioterapia. Desde entonces, se ha precisado extirpación e irradiación con iridio de tres recidivas locales del tumor primario en región periorbitaria izquierda y región frontal bilateral. Un ciclo quimioterápico compuesto de etopósido y cisplatino no resultó efectivo y fue suspendido por la aparición de recidivas locales en el seno del mismo.
COMENTARIO
El tumor de células de Merkel o tumor neuroendocrino cutáneo primario es una entidad infrecuente, descrita por primera vez por Toker en 1972 1, que suele presentarse como una lesión nodular eritematosa de rápido crecimiento en personas mayores de 65 años. Tiene una clara predilección por las zonas fotoexpuestas, localizándose en aproximadamente el 50 % de los casos en cabeza y cuello, seguido, en orden de frecuencia, de extremidades 2. Su curso clínico es a menudo agresivo, con un elevado índice de recurrencias locales (44 %), diseminación linfática regional (55 %) y metástasis a distancia (34 %) 3. La tasa de mortalidad directamente relacionada con el tumor puede alcanzar, según algunas series, el 65 % 4. El tratamiento suele ser multidisciplinar. La resección tumoral se debe llevar a cabo con un margen lateral amplio, de 2 a 3 cm. Debido a la elevada incidencia de metástasis ganglionares regionales clínicamente indetectables en el momento del diagnóstico, algunos autores han recomendado la realización de una linfadenectomía regional electiva, acompañada o no de radioterapia profiláctica sobre las zonas de linfadenectomía y lecho quirúrgico. El tratamiento quimioterápico se suele reservar para casos con metástasis a distancia y/o lesiones recurrentes inoperables 5. El tumor de células de Merkel se ha asociado en numerosos artículos a situaciones de inmunodeficiencia secundaria a neoplasia o, con más frecuencia, a tratamiento inmunosupresor en el contexto de trasplante renal y cardiaco 6. También se ha descrito esta asociación en un paciente con artritis reumatoide de larga evolución en tratamiento con azatioprina 7 y en un paciente seropositivo para el VIH 8. En pacientes inmunocomprometidos con leucemia parece haberse hallado una mayor predisposición a desarrollar tumor de células de Merkel, con un curso más agresivo y una mayor incidencia en edades tempranas de la vida 9.
A pesar de que la gran mayoría de pacientes que desarrollan este tipo de neoplasia cutánea suelen ser diagnosticados bajo la forma clínica anteriormente citada, se han descrito diversas formas de presentación atípicas. Incluyen una mínima ulceración en la punta de la nariz 10, nódulos subcutáneos en región inguinal 11, tejido de granulación en un dedo del pie de un adolescente 12 o una lesión en el pezón de una mujer 13. La presentación a modo de una placa extensa, de coloración vinosa, apenas sobreelevada y de crecimiento rápido, en región frontal, nos llevó a plantear diagnóstico diferencial con un angiosarcoma. El estudio histopatológico, sin embargo, no mostró los característicos canales vasculares revestidos por células endoteliales atípicas propios de esta entidad y fue, en cambio, compatible con un tumor de células de Merkel. Revisando la extensa literatura médica que en las últimas décadas se ha ocupado de definir la clínica variable del tumor de células de Merkel, tan sólo hemos encontrado un artículo que describa un patrón clínico agresivo semejante, a modo de placa erisipeloide, que semeja un angiosarcoma 14.
Conviene llamar la atención, asimismo, sobre la relación que pudiera tener la aparición del tumor de células de Merkel con el estado de inmunosupresión derivado del tratamiento corticoideo prolongado y a las elevadas dosis de la sarcoidosis pulmonar en nuestro paciente.
Por lo tanto, queremos destacar la existencia de esta forma excepcional de presentación clínica de este tumor que semejaba un angiosarcoma. Dado el pronóstico ominoso que presenta esta neoplasia cuando se detecta en estadios avanzados, la importancia de un diagnóstico precoz adecuado se convierte en el principal arma para aumentar la supervivencia de estos pacientes.


