






INTRODUCCIÓN
La pitiriasis rubra pilaris (PRP) es una dermatosis de presentación infrecuente1,2, caracterizada por la aparición de pápulas eritematoescamosas de localización folicular, placas escamosas de color rojo-salmón, que frecuentemente dejan en medio islotes de piel normal, e hiperqueratosis palmoplantar de tonalidad anaranjado-amarillenta1, 3.
Es una enfermedad de gran polimorfismo clínico4. Ello hizo que Griffiths en 19801 la clasificara en cinco tipos diferenciables entre sí, basándose en el aspecto clínico, en la epidemiología y en la evolución (tabla 1). Sin embargo, pese a la reconocida utilidad de esta clasificación, algunos casos son difícilmente encuadrables en ella debido al amplio espectro de formas en que puede presentarse5, 6. Gelmetti et al cuestionaron el pronóstico en el grupo de edad pediátrica, proponiendo en 1986 una clasificación5, 7 basada en la duración de la enfermedad (tabla 2). Como en la mayoría de las enfermedades de etiología desconocida, en la PRP se han empleado múltiples terapéuticas3,4,7. La evaluación de la eficacia de las mismas es difícil debido al curso impredecible de la enfermedad y al carácter, en ocasiones, autoinvolutivo de la misma1,3.


DESCRIPCIÓN DEL CASO
Niña de 5 años, sin antecedentes personales reseñables y sin antecedentes familiares de enfermedad cutánea. Consultó por un cuadro cutáneo de 1 mes de evolución, consistente en la aparición de lesiones rojas y escamosas, secas, a nivel de pulpejos de los dedos de las manos, con progresiva afectación de palmas, plantas, dorso de pies, rodillas y región perioral. No se acompañaba de alteraciones sistémicas y no presentaba fiebre. Había padecido un proceso catarral de vías respiratorias altas 2 semanas antes del inicio del cuadro cutáneo, que no fue tratado farmacológicamente.
La exploración física puso de manifiesto la presencia de queratodermia palmoplantar de color rosado-amarillento, de bordes precisos y descamación laminar (fig. 1A); fisuras en los pulpejos de los dedos de las manos; placas eritematoescamosas en las rodillas, caras laterales, dorso, zonas aquíleas y maleolares externas de ambos pies (fig. 2A). No se apreciaron lesiones en el cuero cabelludo ni en las uñas ni en las mucosas.
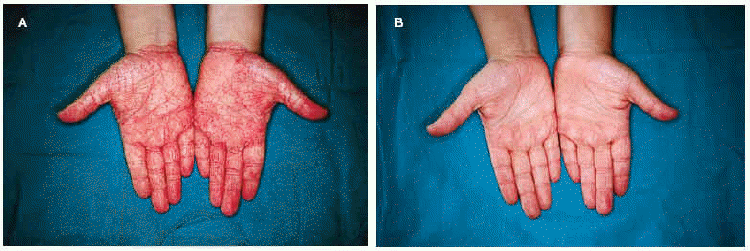
Fig. 1.--A: queratodermia palmar rosada antes del tratamiento. B: tras 2 meses con calcipotriol.
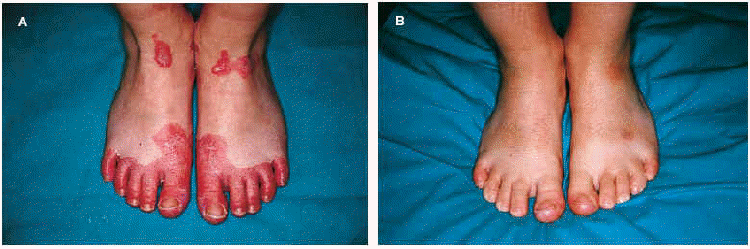
Fig. 2.--A: placas eritematoescamosas en los dedos y el dorso de los pies antes del tratamiento. B: tras 2 meses con calcipotriol.
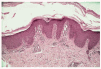
Los estudios analíticos practicados fueron normales (hemograma y bioquímica de sangre y orina). Se realizó una biopsia cutánea a nivel del dorso del pie que mostró hiperplasia psoriasiforme con hiperqueratosis y paraqueratosis focal e hipergranulosis e infiltrado linfocitario perivascular leve en dermis superficial (fig. 3).
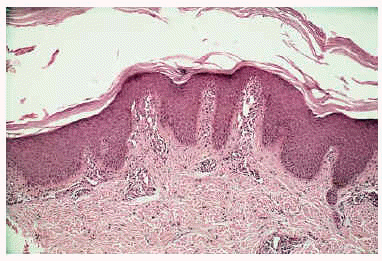
Fig. 3.--Hiperplasia psoriasiforme con hiperqueratosis y paraqueratosis focal.
Establecimos, ante la clínica y los hallazgos histopatológicos sugerentes, el diagnóstico de PRP aguda infantil y comenzamos el tratamiento con calcipotriol crema dos veces al día, presentando buena tolerancia y remisión de las lesiones cutáneas tras 2 meses de tratamiento (figs. 1B y 2B). En la actualidad la paciente presenta únicamente hiperpigmentación postinflamatoria en las zonas lesionales, sin ninguna recidiva tras 7 meses de seguimiento.
DISCUSIÓN
El caso clínico que presentamos no se encuadra en ninguno de los grupos de Griffiths por la presentación y evolución1,7, pero podría ser englobado dentro de la forma denominada PRP aguda infantil postinfecciosa, definida por Larrégue8 en 1983, basándose en cuatro casos propios y 18 observaciones previamente reseñadas en la literatura, individualizándola como entidad, con las siguientes características: ausencia de antecedentes familiares de PRP; aparición durante la infancia, pero no durante el primer año; episodio infeccioso febril previo; comienzo agudo algunos días después del proceso infeccioso; erupción escarlatiniforme seguida de la aparición de pápulas foliculares 2 semanas después; extensión de la erupción que se generaliza en 1 mes, sin llegar a la eritrodermia; aspecto de PRP típica; ausencia de signos clínicos o analíticos asociados4, a no ser los relativos al proceso infeccioso inicial; curación estable, sin recidivas3,8.
La etiología de la PRP sigue siendo desconocida4,9, pareciendo evidente que se trata de un trastorno hiperproliferativo de la queratinización3, habiéndose demostrado elevados índices mitóticos en las células epidérmicas9. Hay diversos factores que pueden contribuir a desencadenar la aparición de este cuadro, como son: radiación solar3, quemaduras químicas3, exposición al frío4, enfermedades hepáticas y diversas infecciones3 (tuberculosis, varicela, gonococia, diversas viriasis2 y sobre todo infecciones faringoamigdalares agudas4). En el caso de los niños parece ser que las infecciones previas desempeñan un papel importante, desencadenando la PRP postinfecciosa3. El vínculo de unión entre la infección y este cuadro hiperproliferativo epidérmico es desconocido3. En cualquier caso parece seguro que esta entidad constituye una forma de respuesta del organismo frente a un estímulo posiblemente antigénico, todavía sin aclarar3.
El diagnóstico de PRP se basa en la correlación clínico-histológica. Los hallazgos histológicos son típicos, pero no patognomónicos, variando según el estadio del proceso9. En la epidermis se aprecia acantosis irregular con hiperqueratosis ortoqueratósica, más marcada en los orificios foliculares donde forma tapones córneos. Es característica la presencia de focos de paraqueratosis en torno al orificio folicular, con ocasional espongiosis y degeneración vacuolar de la capa basal, y a diferencia de la psoriasis la granulosa está conservada o aumentada, no existiendo microabscesos de Munro. En la dermis se observa un infiltrado perivascular mononuclear y atrofia de las glándulas sebáceas, siendo normales los folículos pilosos4,8,10.
El pronóstico de la forma aguda infantil postinfecciosa es muy bueno, con tendencia a la desaparición de la enfermedad de forma espontánea, que ocurrirá en el plazo de unos meses1,4.
Como en la mayoría de las enfermedades cuya causa es desconocida, en la PRP se han utilizado múltiples terapéuticas1,4. La evaluación de la eficacia de las mismas es difícil9, ya que el curso de la enfermedad es impredecible7,11 y en ocasiones autoinvolutivo4,10. Los casos leves son subsidiarios de terapéutica tópica y las formas graves obligan en ocasiones al empleo de tratamientos sistémicos, teniendo en cuenta siempre la expresividad de la enfermedad1,4,12. Nuestro caso fue tratado con calcipotriol tópico, análogo de la vitamina D3, que es efectivo en el tratamiento de la psoriasis13. Sus propiedades son inhibir la proliferación de los queratinocitos y promover la diferenciación de éstos13. Basándose en estas propiedades y en la similitud tanto clínica como histológica entre psoriasis y PRP9,12, van der Kerkhof empleó calcipotriol en un paciente afectado de PRP, obteniendo una buena respuesta clínica. Histológicamente se apreció una reducción del infiltrado de linfocitos T y monocitos, así como reducción en la expresión suprabasal de queratina K1614,15.
Aunque obtuvimos una satisfactoria respuesta terapéutica, con remisión de las lesiones y sin recidivas en 7 meses, es difícil predecir la evolución del proceso en caso de no haber realizado ningún tratamiento dado el carácter autoinvolutivo del mismo. No obstante, pensamos que el calcipotriol tópico es una buena alternativa terapéutica frente a los retinoides sistémicos por su buena tolerancia clínica y por estar exento de la toxicidad sistémica de éstos9,10.


