


INTRODUCCIÓN
La piodermia gangrenosa (PG) es un proceso cutáneo infrecuente que suele manifestarse como úlceras profundas de evolución tórpida en la piel de extremidades inferiores. Aproximadamente el 50% de los casos se asocian a una patología sistémica subyacente (1), entre las que destacan la enfermedad inflamatoria crónica intestinal (EICI), poliartritis, neoplasias hematológicas y paraproteinemias (2-4). Entre los procesos hematológicos, los más frecuentes son las leucemias mieloides y la gammapatía monoclonal IgA. Sin embargo, la leucemia de mayor prevalencia e incidencia en la edad adulta, la leucemia linfática crónica, únicamente se ha descrito asociada a PG en un caso (5). Presentamos el caso de un varón de 72 años que debutó con úlceras rebeldes al tratamiento en extremidades inferiores con diagnóstico concomitante de leucemia linfática crónica. Tras tratamiento con prednisona primero y ciclosporina después, el pioderma gangrenoso presentó buena evolución.
DESCRIPCIÓN DEL CASO
Varón de 72 años sin antecedentes personales de interés que presentó astenia, pérdida de peso, fiebre (hasta 38,5º) y úlceras cutáneas en extremidades inferiores de 4 meses de evolución. La primera de las úlceras había sido tratada con antibioterapia oral y tópica y mediante un injerto cutáneo, pero surgió una nueva lesión en la zona dadora del injerto.
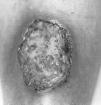
En octubre de 1998 acudió a nuestra Unidad. En la exploración física se apreciaban úlceras de fondo necrótico y borde sobreelevado, eritematoso e infiltrado, con dolor local. Las lesiones se localizaban en cara anterior de muslo derecho y en cara anterior ambas piernas (Fig. 1), alcanzando alguna los 7 cm de diámetro. Se realizó estudio histológico de dos de las lesiones, que mostraban un infiltrado inflamatorio polimorfo, con abundantes histiocitos, células linfoides, plasmáticas y polinucleares, en el seno de un tejido conjuntivo edematoso con marcada proliferación vascular reactiva y ocasionales células gigantes multinucleadas. El aspecto general del infiltrado era inespecífico y no se observaron gérmenes ni granulomas.
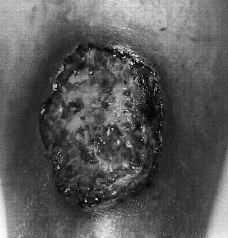
FIG.1.--Úlcera de bordes socavados y fondo necrótico en pierna.
Había también adenopatías submandibulares y axilares bilaterales y laterocervicales y supraclaviculares izquierdas.
En las pruebas complementarias realizadas destacaban: 1) hemograma: hemoglobina, 11,1 g/dl; hematócrito, 37%; leucocitos, 65.800/µl (linfocitos, 78%; segmentados, 14%; monocitos, 8%). Frotis sanguíneo: linfocitos pequeños de aspecto maduro, algunos con sombras nucleares. Inmunofenotipo en sangre periférica: compatible con leucemia linfática crónica B; 2) bioquímica: creatinina, 1,2 mg/dl; ß2 microglobulina, 4,46 mg/dl (normal < 3,75), y 3) ecografía abdominal: esplenomegalia homogénea; TAC cervicotoracoabdominal: múltiples adenopatías en cadenas cervicales, axilares y en grasa mesentérica. Esplenomegalia homogénea.
Las siguientes pruebas fueron normales o negativas: coagulación; proteinograma y cuantificación de inmunoglobulina; serologías VHB, VHC, VIH; crioglobulinas, crioaglutininas y Coombs (directo e indirecto); radiografía de tórax y ecocardiograma; Mantoux.
Con los diagnósticos de leucemia linfática crónica B estadio II de Rai y PG se inició tratamiento con prednisona v.o. a dosis de 1 mg/kg/día. Las lesiones se hicieron menos profundas y disminuyeron de tamaño, pero sin llegar a reepitelizar totalmente. Igualmente desapareció la clínica sistémica, pero el paciente presentó diabetes y miopatía esteroidea, por lo que se redujo progresivamente la dosis de prednisona y se introdujo ciclosporina A a dosis de 4 mg/kg/ día en diciembre de 1998. Las lesiones de PG se resolvieron, dejando cicatrices deprimidas e hiperpigmentadas y se suspendió el tratamiento con ciclosporina A en marzo de 1999. Por progresión de su proceso hematológico en diciembre de 1999 se instaura tratamiento con clorambucil (0,4 mg/kg/día durante 4 días al mes) y esteroides (1 mg/kg/día) con respuesta clínica y analítica. Las lesiones de PG no han recidivado hasta el momento actual.
DISCUSIÓN
La PG es un proceso ulcerativo de etiología desconocida descrito por primera vez en 1930 (6). Sin predominio de sexo, suele aparecer entre los 25 y los 54 años (7). Las lesiones pueden aparecer en cualquier parte del cuerpo (8) (incluidas mucosas), pero su localización más frecuente son las extremidades inferiores y la región abdominal (9). Pueden iniciarse de forma espontánea, pero frecuentemente aparecen tras mínimos traumatismos. Es lo que se denomina fenómeno de patergia. En nuestro paciente quedó bien demostrado al surgir una nueva lesión en la región dadora del injerto cutáneo.
La lesión inicial suele consistir en una pústula que asienta sobre una piel eritematosa, inflamada y dolorosa. La piel central se necrosa y se ulcera y la lesión bien establecida se caracteriza por una úlcera de fondo necrótico, con bordes sobreelevados y bien definidos, eritematovioláceos y socavados.
El diagnóstico es fundamentalmente clínico, puesto que no hay datos de laboratorio concluyentes y la histología no es específica. Los hallazgos histopatológicos son polimorfos, dependiendo fundamentalmente de tres factores: tipo de lesión, tiempo de evolución de la lesión biopsiada y zona de la lesión de donde se tome la muestra (10, 11).
Existen cuatro variantes clínicas e histológicas principales (1): a) PG ulcerativa, variante más frecuente; b) PG pustulosa; suele cursar con fiebre y artralgias; está muy en relación con EICI; se cree que algunos de los casos de dermatosis pustulosa subcórnea de Snnedon-Wilkinson asociados a EICI serían realmente PG (1); c) PG ampollosa o atípica; la clínica y la histología de esta variante, así como su relación con patologías hematológicas, nos ponen en la pista sobre su relación con el síndrome de Sweet. Quizá representen diferentes etapas del mismo espectro patológico (12), y
d) PG vegetativa (o piodermia granulomatosa superficial): son PG superficiales, autolimitadas y crónicas que no suelen asociarse a patología subyacente (13).
Otras variantes más infrecuentes son la piodermia maligna (14-16) o la pyostomatitis vegetans (17).
El 40-50% de las PG se asocian a patología sistémica subyacente (1, 18), predominando la EICI, artritis reumatoidea-like, paraproteinemias y neoplasias hematológicas.
Aproximadamente un tercio de los pacientes con PG asocian EICI (7). En la EICI el 1,5-5% de los pacientes con colitis ulcerosa (CU) y el 1% de los pacientes con enfermedad de Crohn (EC) asocian PG (7), constituyendo la segunda manifestación cutánea más frecuente tras el eritema nodoso. Las variantes ulcerativa y pustulosa son las más frecuentes en esta asociación.
La PG se asocia a procesos artríticos hasta en un 37% de los casos (7), apareciendo normalmente la variante ulcerativa. Suelen ser poliartritis simétricas y seronegativas, que remedan a las de la artritis reumatoidea. También se ha asociado a espondilitis anquilopoyética, artritis reumatoide, enfermedad inflamatoria crónica intestinal con artritis y síndrome de Felty (1).
Un 10% de los pacientes con PG se asocian a gamapatía monoclonal (19), más frecuentemente al tipo IgA, aunque se han descrito asociaciones a tipo IgG e IgM. La asociación de inicio entre un paciente portador de una mieloma y PG es mucho menos frecuente. Estos pacientes suelen presentar la variante ulcerativa.
Otro 7% de los pacientes con PG (fundamentalmente la variante ampollosa) se asocian a procesos linfoproliferativos (20). Se han descrito desde estados preleucémicos (como mielodisplasia) a leucemias bien establecidas (como leucemias mieloides o leucemia linfática aguda), pasando por policitemia vera o linfoma de Hodgkin. Pero sin duda el proceso con mucho más frecuentemente asociado son las leucemias mieloides, y entre ellas la leucemia mieloide aguda (3). Nuestro paciente asociaba a su PG una leucemia linfática crónica (LLC). Esto no debería llamar la atención, puesto que es la leucemia más frecuente entre las que aparecen en la edad adulta (21). Sin embargo, esta asociación es excepcional, habiéndose descrito únicamente un caso en la literatura (5).
La lista de casos aislados de afecciones que se han asociado a PG es muy amplia. Cabe destacar: enfermedad de Behçet, diabetes mellitus, lupus eritematoso sistémico, granulomatosis de Wegener, hepatitis crónica activa, cirrosis biliar primaria, sarcoidosis, hemoglobinuria paroxística nocturna, acné conglobata, hidradenitis supurativa (22) y alteraciones de la inmunidad (como hipogamaglobulinemia o infección por VIH). En Japón casi un tercio de los pacientes con PG presentan asociada vasculitis de grandes vasos (en-fermedad de Takayasu)(23).
Las características clínicas e histológicas de la PG (con el neutrófilo como célula clave), la asociación a patologías hematológicas y digestivas, la ausencia de infección y la respuesta al tratamiento con sulfonas nos sitúa a la PG dentro del espectro de las llamadas dermatosis neutrofílicas y nos plantea la relación estrecha con el síndrome de Sweet, la dermatosis pustulosa subcórnea (24), las erupciones pustulosas generali-zadas de la enfermedad inflamatoria crónica intestinal, el síndrome de Behçet o el erythema elevatum et diutinum (25).
El tratamiento de la PG se basa en el uso de los corticosteroides v.o. (26, 27) y de la ciclosporina A (28-30), aunque se han propuesto múltiples y variados tratamientos. El fenómeno de patergia es una contraindicación relativa al uso de injertos cutáneos, pero existen casos con buena respuesta (31). Siempre que exista una patología asociada, el tratamiento debe ser doblemente dirigido a tratar ambos procesos.


