


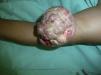

Presentamos el caso de un paciente varón de 7 años de edad que consulta por una lesión localizada en el brazo izquierdo de 2 años de evolución, que había aumentado de tamaño de forma abrupta en el último mes. En la ecografía doppler realizada un mes antes se visualizaba una imagen ecogénica de 1,6×1, 7×1cm con sombra acústica posterior.
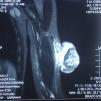
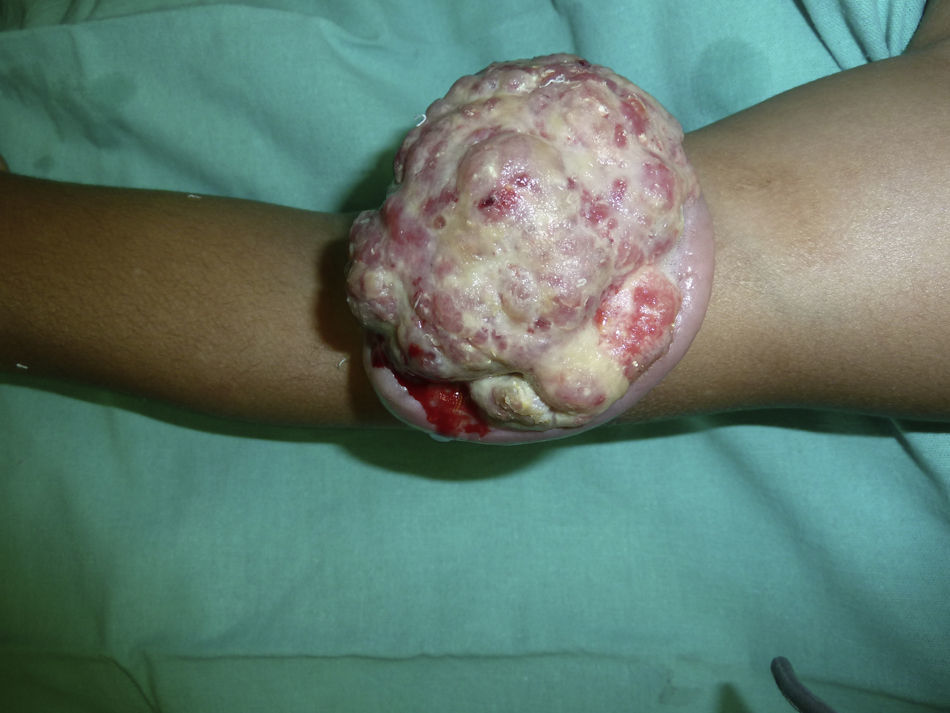
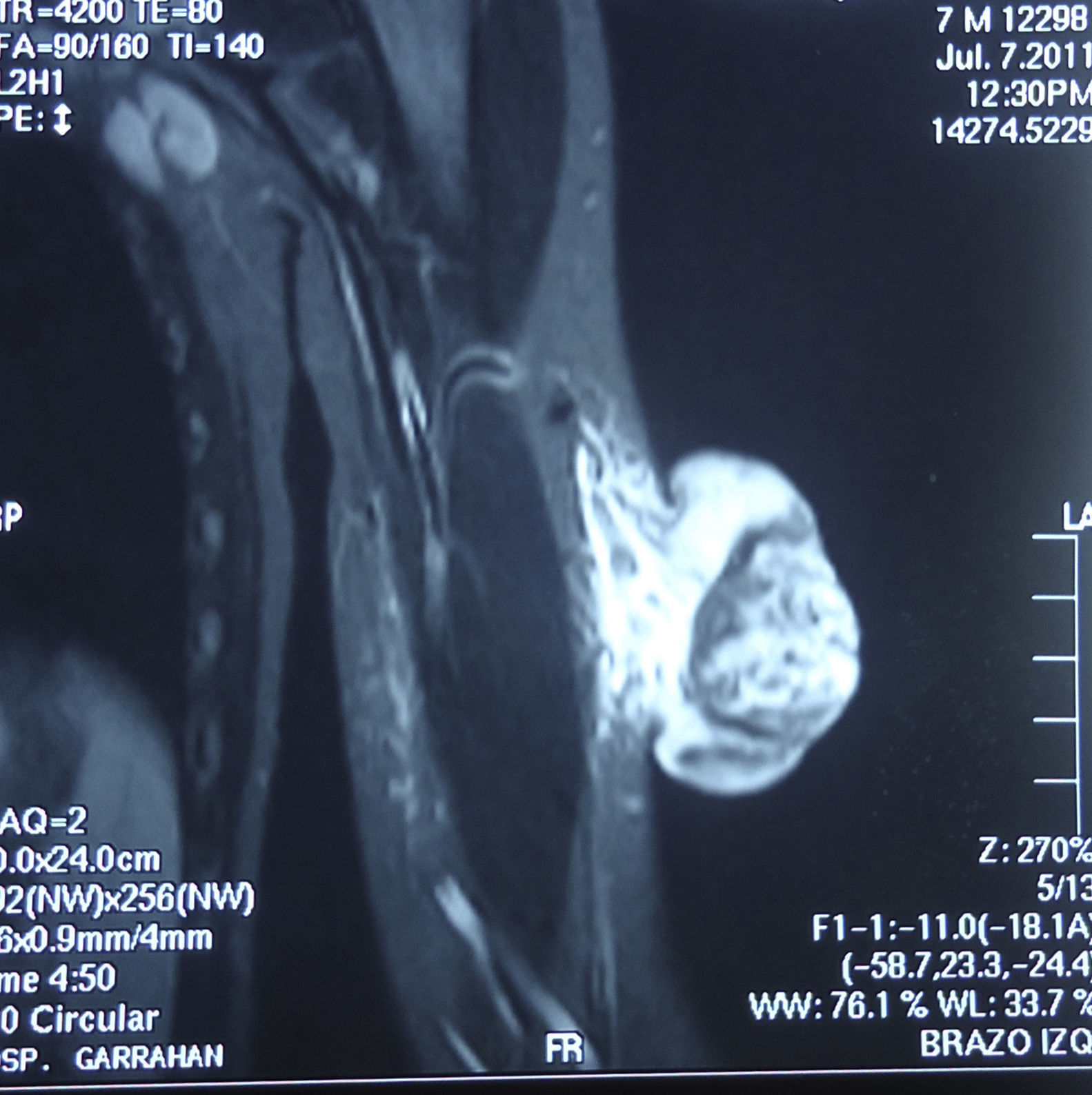
En la exploración se observó en el brazo izquierdo una lesión tumoral exofítica y pediculada, de consistencia pétrea, de 8cm de diámetro, con pérdida de la piel suprayacente, quedando expuesta la superficie erosionada con un lecho sangrante de aspecto crateriforme (fig. 1). Se realizó una resonancia magnética nuclear (RMN) y una angiorresonancia, visualizándose una formación exofítica con un nódulo heterogéneo y un halo hipointenso dentro de un tejido hiperintenso, con gran cantidad de vasos tortuosos en su pedículo (fig. 2). Se realizó exéresis quirúrgica reconstruyendo el defecto mediante un colgajo de Dufourmentel. El estudio histopatológico demostró la presencia de «lóbulos de células basaloides con maduración hacia la luz y formación de células con citoplasma abundante eosinófilo y láminas de detritus queratósicos eosinofílicos y queratinización de tipo pilar. Rodeando los lóbulos el estroma era fibroso con infiltrado inflamatorio mononuclear e histiocitario, con formación de células gigantes multinucleadas tipo cuerpo extraño. Se observaban numerosos focos de calcificación distrófica. Dichos hallazgos eran compatibles con el diagnóstico de pilomatrixoma gigante con reacción gigantocelular de tipo cuerpo extraño» (fig. 3). El paciente evolucionó favorablemente sin recidivas posteriores tras un año de seguimiento.
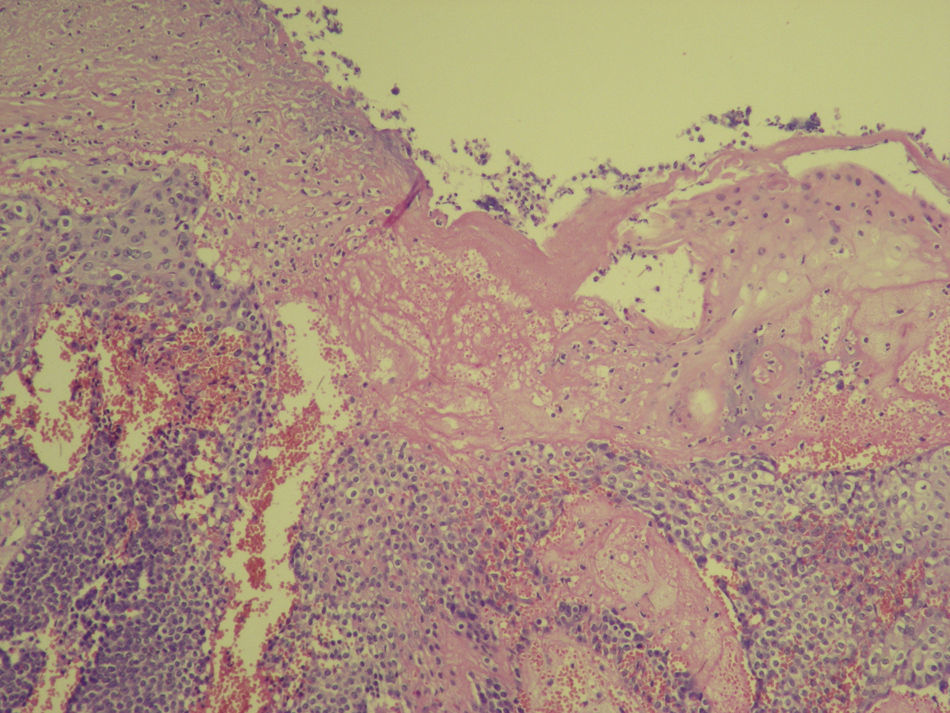
El pilomatrixoma es una neoplasia anexial benigna con diferenciación a células de la matriz del pelo, más frecuente en niños y a partir de la sexta década de la vida1. Aunque de etiología desconocida, en el 75% de los casos se demostró una mutación en el gen de la beta catenina (gen CTNNB1), proteína de señalización involucrada en el desarrollo del folículo piloso2.
Son nódulos que miden entre 1 a 1,5cm, de crecimiento lento que habitualmente se calcifican, dando una coloración azulada a la piel suprayacente, pudiendo extruir material cálcico3. Pueden ser múltiples o solitarios, y estos últimos se han descrito en síndromes genéticos como en síndrome de Turner, síndrome de Sotos, distrofia miotónica, síndrome de Rubinstein-Taybi, síndrome de Gardner, trisomía 9, síndrome del nevo basocelular y xeroderma pigmentoso4. Existen casos de pilomatrixomas múltiples en pacientes sanos5. Se han descrito otras variantes clínico-patológicas como la forma ampollar, anetodérmica, perforante/ulcerada y linfangiectásica o la forma maligna.
Se consideran gigantes los mayores de 5cm. En la actualidad hay publicados solo 4 casos de pilomatrixomas gigantes6–7, 2 de ellos ulcerados y 2 con áreas de erosión, todos en la edad adulta. Hay 2 casos descritos de pilomatrixoma gigante asociados con hipercalcemia que se resolvieron tras la exéresis7.
El diagnóstico diferencial se plantea con el dermatofibrosarcoma protuberans, la calcinosis cutis, el osteoma cutis, los linfomas cutáneos, los sarcomas, los carcinomas espinocelulares y las metástasis cutáneas.
Hay que diferenciar la forma clínica gigante y ulcerada de su forma maligna. Se sospecha ante el crecimiento abrupto de una lesión previa que se ulcera y sangra. El grado y extensión de la infiltración, la necrosis, las figuras mitósicas atípicas, la invasión perineural o perivascular son indicadores de malignidad8.
Como exámenes complementarios la ecografía evidencia una lesión ecogénica con sombra acústica posterior. En este caso la RMN evaluó la extensión y evidenció una formación exofítica con hipervascularización, ya reportada en otro caso de pilomatrixoma gigante9.
El interés de presentar este caso de pilomatrixoma se debe al llamativo y abrupto crecimiento de la lesión, que nos hizo pensar en un probable origen maligno y que se descartó con el resultado de la anatomía patológica.
Se agradece a los siguientes doctores, quienes han participado en la atención y resolución del caso clínico presentado: Dr. Adrián Martín Pierini, Dra. Amelia Laterza y Dra. Fabiana Lubieniecki, del Hospital Nacional de Pediatría Prof. Dr. JP Garrahan.


