

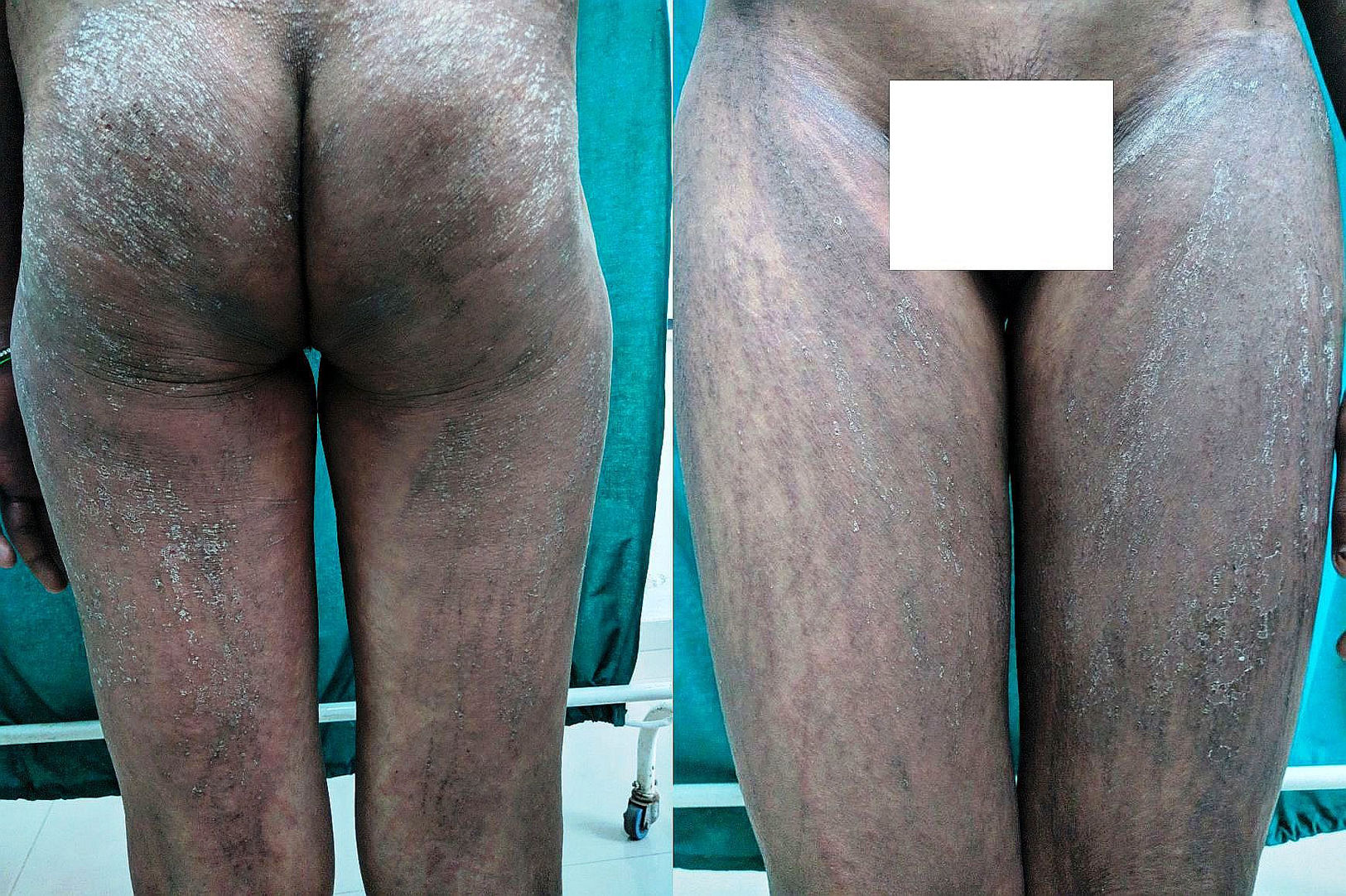
A 21-year-old lady presented to our hospital with an 8-month history of persistent polyarthralgias, sore throat, intermittent fevers, and intense pruritic skin lesions over her face, upper back and chest, buttocks and extremities. Her past and family history was unremarkable and there was no history of any drug intake prior to onset of these symptoms. Cutaneous examination revealed erythematous and hyperpigmented, slightly scaly excoriated papules over forehead, upper back and chest and similar lesions in a linear manner over her buttocks and upper and lower extremities (Fig. 1). Laboratory investigations revealed leukocytosis (16,800/uL, 90% neutrophils) and elevated erythrocyte sedimentation rate [ESR] (40mm/hr), ferritin (12,000ng/mL) and C-reactive protein (8mg/dL). Antistreptolysin titers, antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibodies, serum creatine phosphokinase, and aldolase were negative or within normal limits. Blood and urine cultures and other investigations for hepatitis B, C and A viruses, and chikungunya were all negative. Chest radiograph, echocardiogram, ultrasonography of abdomen and pelvis, and electromyogram did not reveal any abnormality.
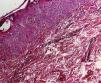
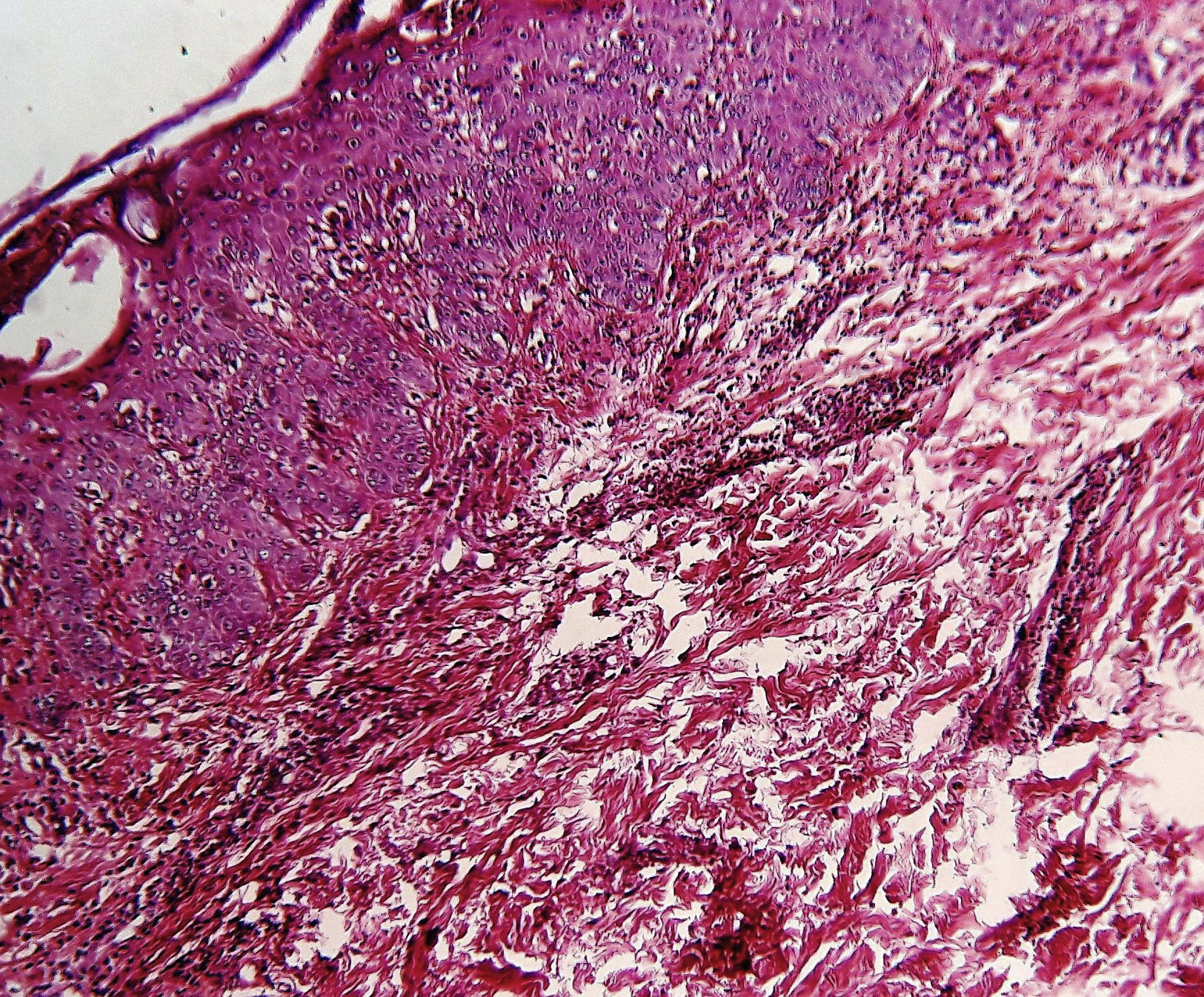
Histological examination revealed microabscesses in the stratum corneum with necrotic keratinocytes and perivascular and periadnexal inflammatory infiltrate (Fig. 2). Considering her clinical and histological findings, diagnosis of adult onset Still's disease (AOSD) was made and she was treated with oral steroid (1mg/kg/day). There was significant resolution of her cutaneous lesions and other symptoms also ameliorated at the 4 weeks of follow-up.
Still's disease is an idiopathic systemic inflammatory disorder with seronegative arthritis. Patients with 16 years of age or older having Sill's disease are labeled as AOSD, whereas younger than 16 years are termed as Juvenile Still's disease.1 Yamaguchi et al.2 have led the major diagnostic criteria to diagnose AOSD, which include high spiking fevers ≥39°C for at least 1 week, leukocytosis with neutrophilia, arthralgia for more than 2 weeks, and typical skin eruption; whereas the minor criteria include sore throat, lymphadenopathy, splenomegaly, hepatic dysfunction, negative rheumatoid factor and antinuclear antibody. AOSD is diagnosed when five or more criteria (including 2 major criteria) are present in the patient, provided there is no evidence of infections, malignant conditions, and other rheumatologic diseases. However Sun NZ et al. have found the delayed association of malignancies (breast cancer and lymphoma) with AOSD, especially in patients having atypical cutaneous lesions.3
The classic rash of AOSD consists of an evanescent, non-pruritic salmon pink, morbilliform eruption on the trunk and/or extremities which appears simultaneously with fever spikes. The non-classic variant of the skin rash in AOSD which is characterized by pruritic papules and plaques with fine scales along with flagellate erythema located on the trunk, extremities, head and/or neck. Owing to its itchy nature, the linearity of this rash over especially over extremities may represent a Koebner phenomenon. Flagellate erythema have been observed in patients undergoing chemotherapy with bleomycin, peplomycin, docetaxel and transtuzumab and also in patients suffering from dermatomyositis, systemic lupus erythematosus, chikungunya fever and Parvoirus B19 infection and after the consumption of shiitake mushroom.4
The histopathologic findings of AOSD are nonspecific, which include mild superficial perivascular lymphocytic infiltrate with variable neutrophils, whereas the histopathology of atypical rash of AOSD include dyskeratosis or parakeratosis and necrotic keratinocytes in the upper layers of the epidermis extending into the stratum corneum and a sparse superficial dermal infiltrate containing neutrophils without vasculitis. In some cases dermal mucin deposits are present.5 Erythema multiforme is the prototype diagnosis within the histologic differential of necrotic keratinocytes, which also includes Stevens-Johnson syndrome, toxic epidermal necrolysis, lichen planus, lupus, and graft-versus-host disease. However in EM the necrotic keratinocytes are scattered throughout the epidermis, whereas in Still's disease they are situated in the upper epidermis.5
Other atypical skin lesions less commonly seen in AOSD include urticaria and urticaria-like eruptions, generalized non-pruritic persistent erythema, vesiculopustular eruptions, a widespread peau d’orange appearance of the skin and edema of the eyelids mimicking dermatomyositis.6,7 Recently few atypical forms of AOSD reported include neutrophilic urticarial dermatosis,8 brown macules on oral mucosa9 and generealised purpura.10
The majorities of patients of AOSD with atypical cutaneous lesions have high levels of ferritin and they often suffer from persistent and severe disease.3 Most patients respond to medium or high doses of glucocorticoids while those with severe and persistent disease need a more potent immunosuppressant drugs including methotrexate, azathioprine, cyclosporine A, hydroxychloroquin and IL-1 receptor antagonist.1
Treating physician should be aware of the atypical cutaneous features of AOSD as it is a poor prognostic marker needing prompt diagnosis and aggressive treatment measures.
FundingThis work has not received any type of funding.
Conflict of interestThe authors declare that they have no conflict of interest.
Authors want to thank Dr. Asmita More, MD (Medicine), Dept. of Medicine, Dr. Vasantrao Pawar Medical College & Hospital & Research Center, Nashik, India for providing valuable inputs in this case.
Please cite this article as: Pawar M, Zawar V, Kumavat S. Pápulas y placas pruriginosas persistentes y eritema flagelado como manifestaciones de una enfermedad de Still del adulto. Actas Dermosifiliogr. 2020. https://doi.org/10.1016/j.ad.2019.01.031


