




















El penfigoide ampolloso (PA) es una enfermedad ampollosa autoinmune causada por anticuerpos dirigidos contra componentes de la membrana basal. La mayoría de estos anticuerpos son de clase IgG y se unen principalmente a 2 proteínas hemidesmosómicas, los antígenos BP180 y BP230.
Se trata de la enfermedad ampollosa más frecuente en los países desarrollados en la población adulta, con una incidencia estimada en nuestro medio de 0,2 a 3 casos nuevos por cada 100.000 habitantes. Afecta principalmente a pacientes ancianos, aunque puede presentarse en jóvenes e incluso niños.
En los últimos años hemos incorporado a la práctica clínica nuevas técnicas diagnósticas (ELISA para BP180) y nuevos fármacos con distintas dianas terapéuticas en el tratamiento del PA. Esto ha permitido un avance en el conocimiento y el manejo de esta entidad.
A pesar de ello, no existen a día de hoy unas guías consensuadas internacionalmente sobre el manejo del PA.
Este artículo es una revisión actualizada de la literatura científica sobre el tratamiento de esta enfermedad, destacando las recomendaciones basadas en la evidencia y desde un punto de vista práctico basado en la experiencia del manejo diario de estos pacientes.
Bullous pemphigoid (BP) is an autoimmune subepidermal bullous disease in which autoantibodies are directed against components of the basement membrane. Most of these antibodies belong to the immunoglobulin G class and bind principally to 2 hemidesmosomal proteins: the 180-kD antigen (BP180) and the 230-kD antigen (BP230). It is the most common blistering disease in the adult population in developed countries, with an estimated incidence in Spain of 0.2 to 3 cases per 100,000 inhabitants per year. The disease primarily affects older people, although it can also occur in young people and even in children. In recent years, advances in clinical practice have led to a better understanding and improved management of this disorder. These advances include new diagnostic techniques, such as enzyme-linked immunosorbent assay for BP180 and new drugs for the treatment of BP, with diverse therapeutic targets. There is, however, still no international consensus on guidelines for the management of BP. This article is an updated review of the scientific literature on the treatment of BP. It focuses primarily on evidence-based recommendations and is written from a practical standpoint based on experience in the routine management of this disease.
El penfigoide ampolloso (PA) es una enfermedad ampollosa autoinmune en la que se producen anticuerpos dirigidos contra componentes de la membrana basal. La mayoría de estos anticuerpos son de clase IgG y se unen principalmente a 2 proteínas hemidesmosómicas, los antígenos BP180 (también denominado colágeno xvii) y BP230. Existen evidencias experimentales y clínicas de que estos autoanticuerpos (principalmente los dirigidos contra BP180) son los responsables de la formación de las ampollas, y por lo tanto los causantes de la enfermedad.
Disponemos de varios artículos recientes que recogen interesantes recomendaciones sobre el tratamiento de estos pacientes1–4, sin embargo no existen a día de hoy unas guías consensuadas internacionalmente sobre el manejo del PA ni disponemos de guías actualizadas en castellano.
En este artículo hemos realizado una revisión actualizada de la literatura científica sobre el tratamiento de esta enfermedad, destacando las recomendaciones basadas en la evidencia y desde un punto de vista práctico basadas en la experiencia del manejo diario de estos pacientes.
EpidemiologíaEl PA es la enfermedad ampollosa autoinmune más frecuente en los países desarrollados en la población adulta. Su incidencia estimada en nuestro medio es de 0,2 a 3 casos nuevos por cada 100.000 habitantes. Algunos estudios recientes apuntan a un incremento de estas cifras en los últimos años5, aunque en un estudio muy reciente en Francia se ha encontrado una incidencia de 21,7 casos por millón de personas al año6. La incidencia es similar en hombres y mujeres. La mayoría de los casos se presentan en personas mayores de 75 años, pero puede afectar a jóvenes e incluso a niños. Se han descrito diversos casos de PA asociado a distintas neoplasias, si bien estudios realizados con controles emparejados por sexo y edad no han observado un aumento de enfermedades malignas en los pacientes con PA7.
PatogeniaLa patogenia del PA está definida básicamente por 2 componentes:
- 1.
Inmunológico: determinado por la presencia de anticuerpos frente a proteínas de los hemidesmosomas de los queratinocitos basales (principalmente los antígenos BP180 y BP230).
- 2.
Inflamatorio: tiene mayor importancia que en otras enfermedades ampollosas autoinmunes. Este componente se halla determinado por la acción de polimorfonucleares (neutrófilos, eosinófilos), que serían activados por la fracción Fc de los autoanticuerpos, produciendo la liberación de enzimas proteolíticas que dañarían la unión dermo-epidérmica.
Hasta hace poco se consideraba a las IgG anti-BP180 como los únicos anticuerpos patógenos en el PA. Sin embargo, se ha demostrado que estos pacientes presentan, además, anticuerpos tipo IgE específicos contra el BP180, y estudios recientes con modelos animales apuntan a un papel patogénico de estos anticuerpos en la formación de lesiones cutáneas de PA8–10, así como a una posible correlación de sus títulos con la actividad de la enfermedad, tal y como sucede con los anticuerpos IgG anti BP 18011.
Una larga lista de fármacos (espironolactona, furosemida, bumetanida, D-penicilamina, amoxicilina, ciprofloxacino, yoduro potásico, sales de oro, captopril) se ha relacionado con el desencadenamiento de PA, aunque desconocemos el mecanismo preciso por el que esto ocurre. Se ha planteado la hipótesis de que estos fármacos puedan modificar la respuesta inmune o bien alterar los antígenos de la membrana basal en pacientes con una predisposición genética determinada3. Además se han descrito casos de PA inducidos por fototerapia, radioterapia, vacunas e infecciones víricas, así como en el contexto de un rechazo agudo o crónico a un trasplante. La detección de estos posibles factores desencadenantes depende en gran medida de la realización de una buena historia clínica, y es especialmente importante pensar en ello ante pacientes ancianos, generalmente expuestos a numerosas medicaciones. El control o eliminación del factor desencadenante, siempre que sea factible, puede facilitar de forma decisiva el manejo de algunos pacientes con PA.
ClínicaEl PA se caracteriza clínicamente por la aparición inicial de lesiones urticariformes o eccematosas muy pruriginosas sobre las que pueden aparecer al cabo de un tiempo variable ampollas tensas (fig. 1). En ocasiones cursa exclusivamente con prurito intenso, y las únicas lesiones apreciables son excoriaciones, por lo que el penfigoide debe tenerse en cuenta ante cualquier cuadro de prurito crónico en ancianos. Inicialmente aparecen vesículas, pero posteriormente las lesiones pueden transformarse en ampollas que llegan a alcanzar un tamaño grande y suelen tener un contenido seroso o hemorrágico. Se localizan principalmente en el tronco y en la superficie flexora de las extremidades, siendo infrecuente que se afecte la cabeza y el cuello. El signo de Nikolsky es negativo, aunque su presencia no excluye el diagnóstico (los autores han observado algún caso de PA con positividad en este signo).
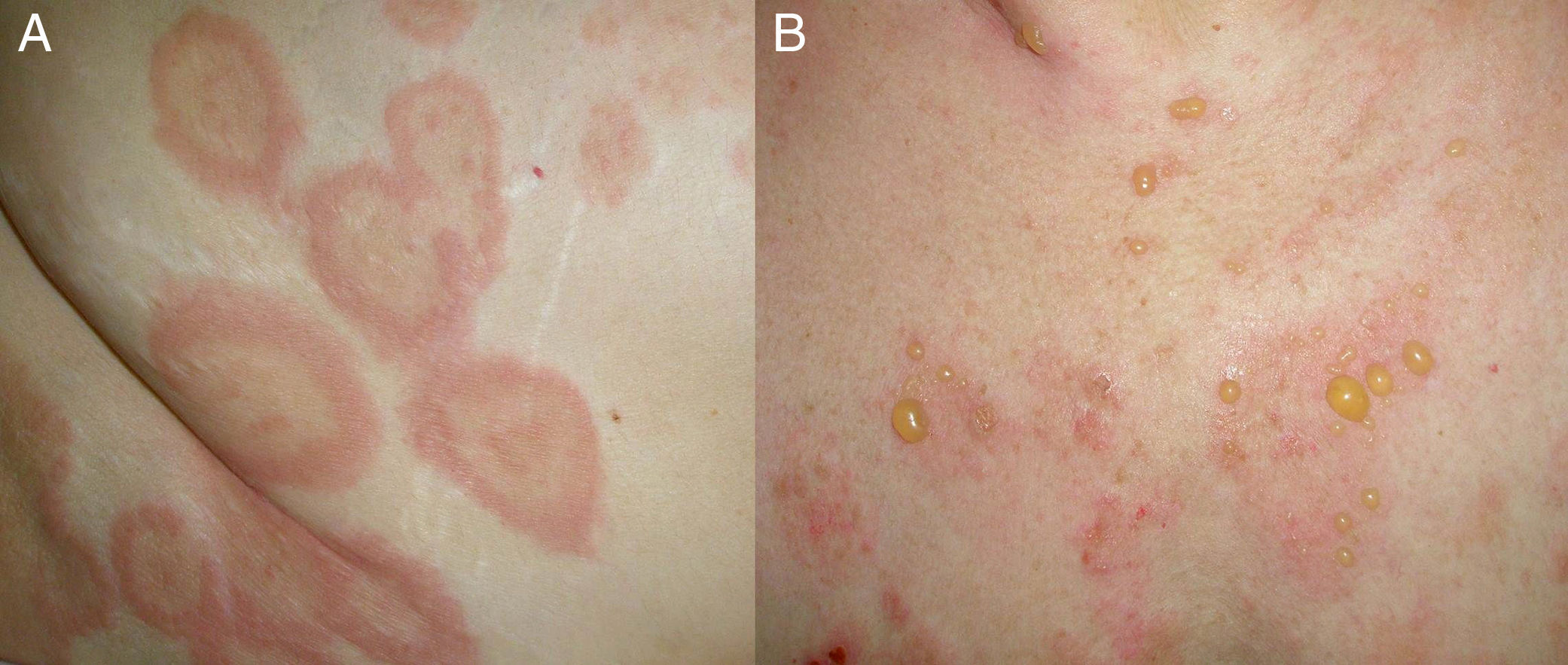
Las ampollas se resuelven sin dejar cicatriz, a menudo dejan una hiperpigmentación postinflamatoria. Existen casos de formas localizadas y atípicas de PA que incluyen: penfigoide dishidrótico (afectación palmoplantar), penfigoide eritrodérmico, penfigoide nodular (similar a un prurigo nodular), formas limitadas a una zona de piel irradiada, liquen plano penfigoide, penfigoide vegetante, etc. La afectación mucosa es rara y cuando se produce suele ser leve y generalmente limitada a la mucosa oral.
DiagnósticoPara realizar un diagnóstico de PA debemos basarnos en los datos clínicos, histológicos e inmunológicos. No existen criterios diagnósticos establecidos como tales, a diferencia de otras enfermedades, aunque un estudio del grupo francés describió los siguientes criterios clínicos de PA a considerar ante un paciente con una erupción ampollosa: ausencia de cicatrices atróficas, ausencia de afectación de cabeza y cuello, ausencia de afectación de mucosas y edad superior a 70 años. La presencia de 3 de estos 4 criterios presentaría una sensibilidad del 90%, una especificidad del 83% y un valor predictivo positivo del 95%12.
Ante la sospecha diagnóstica de PA deben realizarse 2 biopsias cutáneas:
- 1.
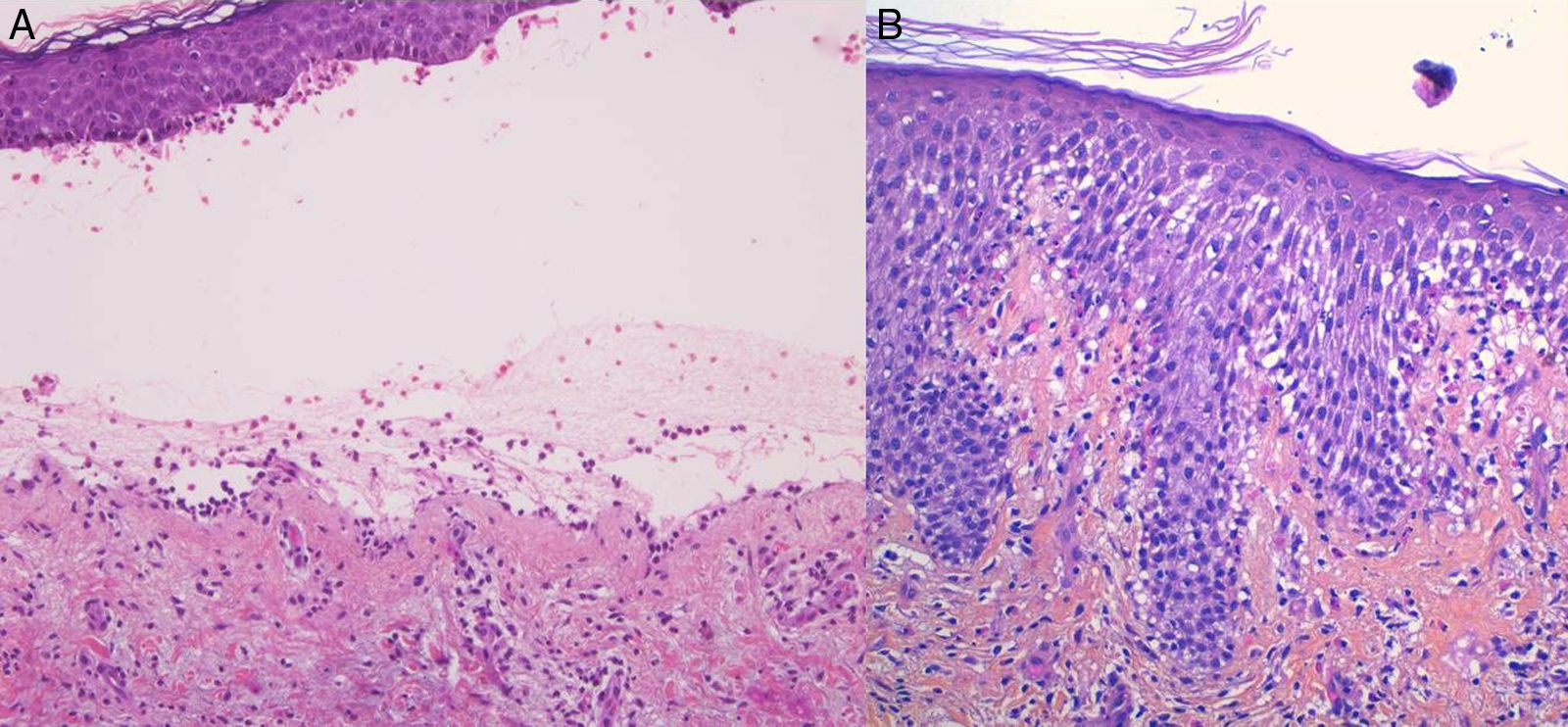
Para el estudio histológico estándar se tomará una muestra de una lesión vesiculosa reciente (para evitar fenómenos de reepitelización) y se incluirá en formaldehído. Se aprecia típicamente una ampolla subepidérmica con un infiltrado inflamatorio perivascular superficial mixto con abundantes eosinófilos (fig. 2A). No se observan imágenes de vasculitis, y los infiltrados inflamatorios no suelen afectar la dermis reticular profunda o el panículo (a diferencia de lo que sucede, por ejemplo, en las reacciones a picaduras de artrópodo). En las lesiones iniciales o fases preampollosas es característico observar el patrón de espongiosis eosinofílica (fig. 2B). A pesar de que no es diagnóstico, la presencia de espongiosis eosinofílica en un paciente de más de 70 años, con presencia de eosinófilos alineados a lo largo de la membrana basal es muy sugestiva de PA.
- 2.
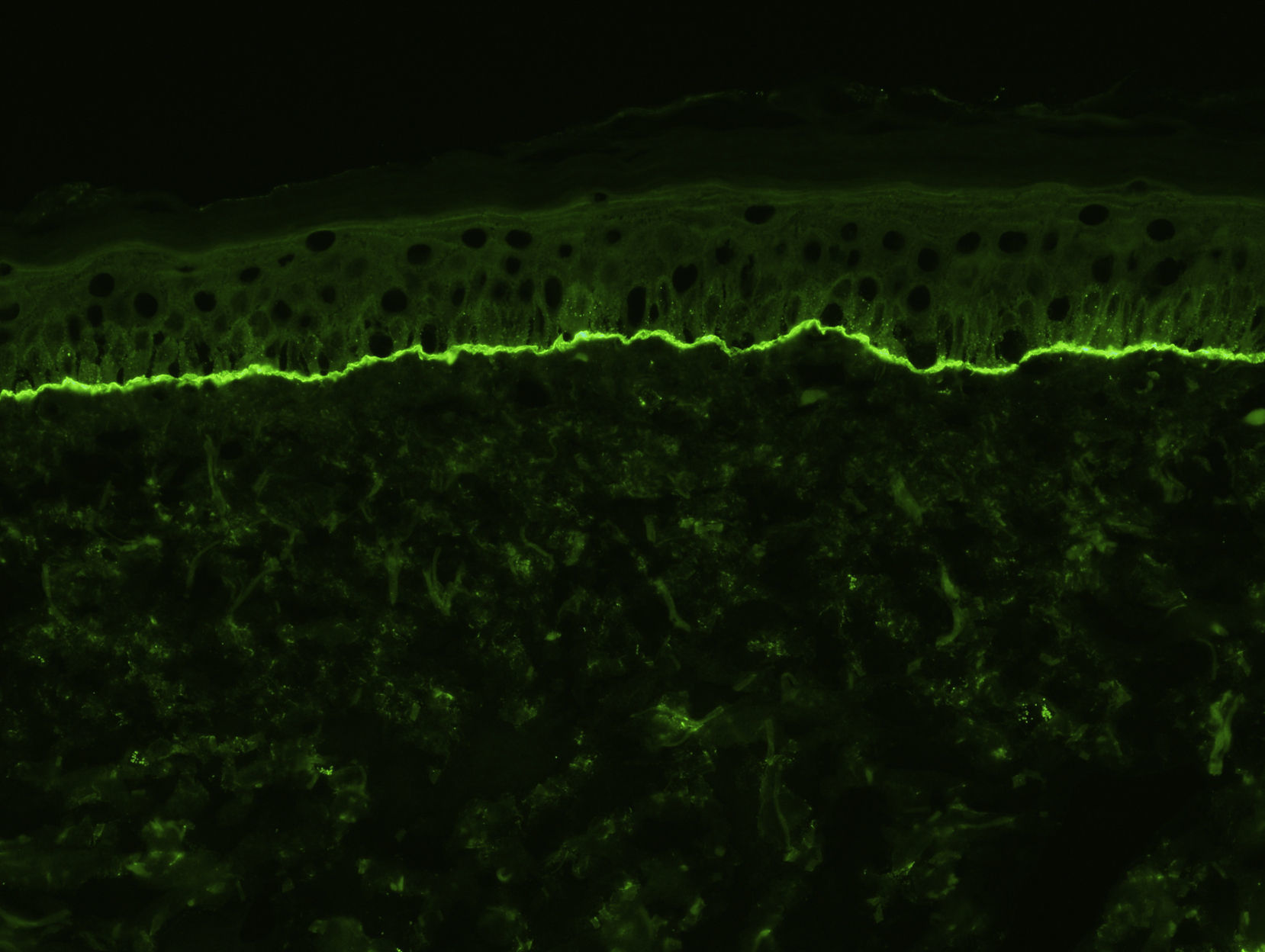
Para el estudio de inmunofluorescencia directa (IFD) se obtendrá otra muestra de piel sana perilesional o de piel inflamada pero sin vesículas ni ampollas (para evitar los falsos negativos que pueden aparecer cuando existen vesículas). Se puede recoger en fresco (para congelar inmediatamente), en suero fisiológico, o en medio de Michel. Se ha podido demostrar que los mejores resultados se obtienen con biopsias conservadas en suero fisiológico durante 12-24h13. En el caso de biopsias que puedan tardar más de 24h en llegar al laboratorio la pieza debería remitirse en medio de transporte de Michel, que permite conservarla durante varias semanas, incluso a temperatura ambiente14. En la IFD característicamente se observan depósitos lineales de IgG y C3 en la membrana basal, aunque puede haber también depósitos de otras inmunoglobulinas, como IgM o IgA, pero siempre de menor intensidad (fig. 3). Típicamente los depósitos de C3 son más intensos que los de IgG (si observamos depósitos mucho más intensos de IgG esto nos debe hacer pensar en otras enfermedades, como la epidermólisis ampollosa adquirida). En un pequeño porcentaje de pacientes con PA se observan depósitos exclusivamente de C3, sin presencia de IgG. Otro hallazgo descrito como característico del PA es que los depósitos de IgG adoptan un patrón en «N serrada» (a diferencia de la epidermólisis ampollosa que serían en «U serrada»), aunque en nuestra experiencia este hallazgo es difícil de objetivar15.
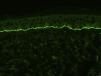
La inmunofluorescencia indirecta (IFI) nos permite estudiar la presencia de los anticuerpos anti-membrana basal circulantes en sangre periférica o en el contenido de una ampolla. Esta técnica puede realizarse utilizando como sustrato esófago de mono, o mejor empleando piel humana tratada mediante una solución de cloruro sódico 1M (técnica del salt split, que permite separar la membrana basal en el plano de la lámina lúcida) (fig. 4). Esta última presenta la ventaja de tener una mayor sensibilidad, y además permite diferenciar los anticuerpos del PA (que se depositan principalmente en el lado epidérmico de la ampolla creada artificialmente) de otras enfermedades como la epidermólisis ampollosa adquirida. En algunos pacientes en los que la IFI es negativa se puede realizar una técnica de IFD separando también la biopsia mediante la técnica del salt split16.
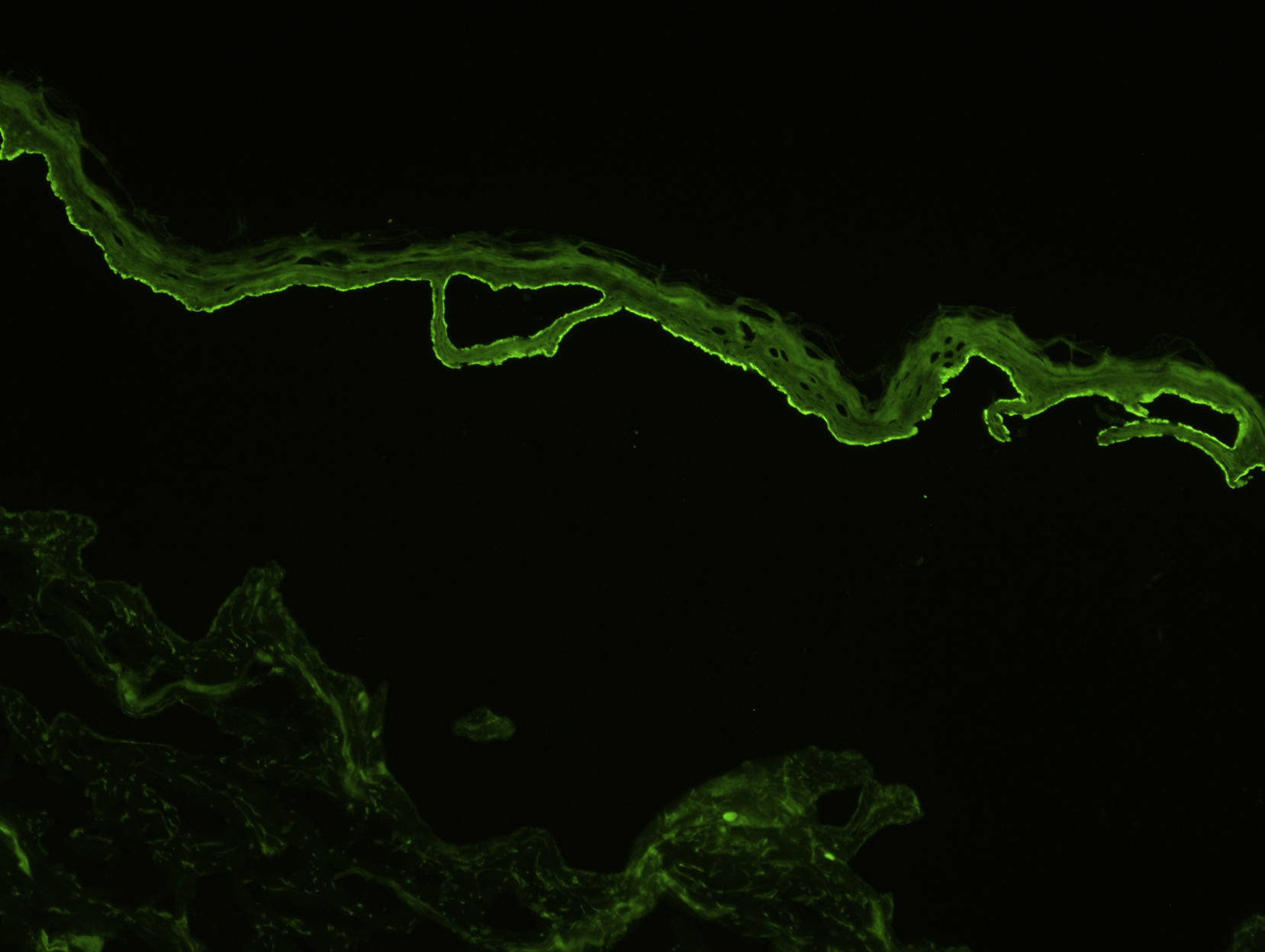
Por último, existen varias técnicas para determinar la especificidad antigénica de los autoanticuerpos. Algunas, como el inmunoblot, permiten observar la presencia de anticuerpos contra proteínas de 180 y/o 230kD, que corresponden a los antígenos BP180 y BP230 (cuando se emplean extractos epidérmicos), o la presencia de anticuerpos contra la porción NC16A del antígeno BP180 del PA (cuando se emplean proteínas recombinantes). Estas técnicas, sin embargo, solo se utilizan en laboratorios muy especializados y con fines de investigación. En la actualidad la más empleada es la técnica de ELISA, que consigue detectar anticuerpos circulantes contra los 2 antígenos del PA (BP180 y BP230) en un gran porcentaje de pacientes. La determinación cuantitativa es útil para monitorizar a los pacientes, dado que se ha demostrado que los niveles de anticuerpos anti-BP180 se correlacionan con la actividad clínica. Dicha técnica está comercializada por 2 laboratorios: Euroimmun, Lubeck (Alemania) y MBL, Nagoya (Japón). Finalmente, en los últimos años el laboratorio Euroimmun, junto con el departamento de Dermatología de la Universidad de Lübeck (Alemania) está poniendo a punto biochips tisulares que utilizan un panel de sustratos, que incluyen esófago de mono, piel humana separada con cloruro sódico 1M, células transfectadas con diferentes antígenos y agregados proteicos que se pueden utilizar como sustrato de IFI y permiten la identificación directa de los antígenos diana del PA y de otras enfermedades ampollosas.
Diagnóstico diferencialLas enfermedades ampollosas del grupo de los pénfigos suelen diferenciarse clínicamente por la presencia de ampollas frágiles, que se rompen fácilmente, y con un signo de Nikolsky positivo. La histología (ampolla intraepidérmica en los pénfigos, subepidérmica en el PA) y los estudios inmunológicos confirmarán el diagnóstico.
El PA debe diferenciarse sobre todo de otras enfermedades caracterizadas por la presencia de ampollas subepidérmicas como la dermatitis herpetiforme, la epidermólisis ampollosa adquirida, la dermatosis IgA lineal, el penfigoide de mucosas u otras enfermedades aún más infrecuentes (como el penfigoide anti-laminina 332, o el penfigoide anti-laminina gamma-1) en función de la clínica, de la histología y de los hallazgos de los estudios de IFD e IFI. En algunos pacientes puede ser imprescindible realizar la determinación de la especificidad antigénica de los autoanticuerpos.
Otras enfermedades como urticaria, eczema, urticaria vasculitis, eritema multiforme, toxicodermias, escabiosis o algunos exantemas víricos (especialmente en adultos) pueden semejarse clínicamente a las lesiones iniciales de PA. El estudio histológico, inmunológico y diversos datos analíticos o de cultivo hacen posible el diagnóstico diferencial.
PronósticoEl PA suele ser una enfermedad autolimitada, aunque puede durar varios años, generalmente menos de 5. La mortalidad previa al uso de corticoides para su tratamiento era del 24%17. Los datos de los que disponemos sobre la mortalidad actual, según distintos estudios, varían ampliamente desde el 6 al 40%. En un estudio reciente de Francia, el más amplio publicado hasta la actualidad, que incluía 502 pacientes, se encontró una mortalidad en el primer año del 38%, siendo la mortalidad 6 veces mayor que la población general6. Estos datos contrastan con nuestra propia experiencia, dado que en una revisión realizada sobre 101 pacientes de nuestro centro encontramos una mortalidad en el primer año del 13%, siendo la edad avanzada (más de 80 años) el único factor de riesgo para predecir la mortalidad18 (Gual et al. Manuscrito sometido a publicación).
La presencia de prurito afecta de forma importante a la calidad de vida de los pacientes. Por otro lado, las lesiones erosivas representan una posible puerta de entrada para una infección, y si son extensas predisponen al paciente a una considerable pérdida de líquidos y electrolitos, así como a una alteración de la termorregulación. Debemos tener en cuenta que generalmente el PA ocurre en personas de edad avanzada, frecuentemente con comorbolidades y polimedicadas, y que son por tanto pacientes frágiles, con mayor riesgo de presentar complicaciones y efectos secundarios a la medicación.
Estudios complementariosEn los pacientes con sospecha o diagnóstico confirmado de PA se debe solicitar diversas pruebas complementarias que se realizarán preferiblemente antes de iniciar el tratamiento:
- 1.
Analítica con hemograma completo, bioquímica básica, perfil hepático y renal.
- 2.
Serologías víricas: VHC, VHB y VIH, VVZ.
- 3.
Actividad de la enzima tiopuril metil transferasa (TPMT) en el caso de administrar azatioprina.
- 4.
Actividad de la enzima glucosa 6 fosfato deshidrogenasa (G6PDH) en el caso de administrar sulfona.
- 5.
Toma de presión arterial.
- 6.
Radiografía de tórax.
- 7.
Test de Mantoux (PPD).
Los fármacos utilizados en el tratamiento del PA presentan 3 mecanismos básicos de acción:
- 1.
Fármacos antiinflamatorios como los corticoides tópicos, la sulfona, las sulfamidas o antibióticos con propiedades antiinflamatorias como las tetraciclinas.
- 2.
Fármacos cuyo objetivo es disminuir la producción de anticuerpos patógenos como los corticoides sistémicos, azatioprina, micofenolato, ciclofosfamida, metotrexato, ciclosporina o rituximab.
- 3.
Otros tratamientos que aumentan la eliminación de anticuerpos patógenos del suero de los pacientes como las inmunoglobulinas endovenosas a dosis altas o la plasmaféresis.
Aunque no existen guías de consenso sobre el manejo de los pacientes con PA19, a la hora de pautar un tratamiento debemos considerar que se trata de una enfermedad con elevada morbilidad (aunque inferior a otras enfermedades ampollosas como el pénfigo vulgar) y que afecta principalmente a personas de edad avanzada.
En general es preferible emplear fármacos lo menos tóxicos posible, e intentar actuar sobre el componente inflamatorio de la enfermedad20. Debemos tener en cuenta que, al igual que en otras enfermedades autoinmunes, el tratamiento en el PA no es curativo, sino que solo suprime la actividad de la enfermedad. En caso de presentación localizada la corticoterapia tópica de alta potencia puede ser suficiente para controlar la enfermedad, y es por tanto el tratamiento de elección21,22.
Consideraciones prácticasEn la práctica la elección del tratamiento y el manejo general dependerá en gran medida del tipo de paciente ante el que nos encontramos. Al enfrentarse a grupos específicos de pacientes, como son los pacientes de edad avanzada o aquellos en edad pediátrica, deben tenerse en cuenta algunas consideraciones.
Pacientes de edad avanzadaSuelen presentar un sistema inmunológico ya deteriorado debido a la edad y las comorbilidades, por lo que la recomendación de evitar el uso de fármacos inmunosupresores cobra aún mayor interés.
En estos pacientes es importante evaluar y tener en cuenta su capacidad o dependencia para las actividades cotidianas, así como su entorno (vive con su familia, está institucionalizado…) y el apoyo o ayuda de la que dispone, que pueden determinar el tratamiento a indicar (por ejemplo, le resultará difícil realizar un tratamiento tópico a una persona con movilidad reducida y sin ayuda). Existen escalas como la escala Karnofsky (desarrollada para pacientes con cáncer) que valora el estado general del paciente y puede orientar esta evaluación.
Por otro lado, las comorbilidades y el uso de fármacos tan frecuentes en los pacientes de edad avanzada deben ser recogidas cuidadosamente en la historia clínica inicial, así como los posibles cambios durante el curso de la enfermedad, con el objetivo de evitar el uso de fármacos contraindicados, interacciones farmacológicas, etc.
Debemos recordar además que las funciones cognitivas de estos pacientes pueden estar disminuidas o alteradas. En estos casos es difícil valorar ciertos aspectos clínicos, como el prurito o la tolerancia a los fármacos, así como dar instrucciones sobre el tratamiento. En nuestra experiencia es importante informar adecuadamente, apoyar e implicar a los familiares o personas encargadas del cuidado del paciente en el control de la enfermedad.
Pacientes en edad pediátricaEl PA infantil presenta típicamente mejor pronóstico y una evolución menos agresiva que en adultos23. En estos pacientes es importante minimizar el empleo de fármacos tóxicos y de corticoides sistémicos debido a sus efectos secundarios, especialmente la posible interacción en el crecimiento y desarrollo del paciente. En el caso de utilizar corticoides sistémicos es recomendable recoger los datos en cuanto a talla y percentiles del paciente de forma inicial para poder controlar posibles cambios a lo largo del seguimiento. En nuestra experiencia hemos observado casos de PA infantil con un curso agresivo y difícil manejo que han requerido fármacos inmunosupresores y corticoides sistémicos para su control. Consideramos de gran utilidad que el paciente pediátrico en tratamiento sistémico sea controlado de forma multidisciplinar junto a especialistas en Pediatría. Es importante recordar que la retirada de los corticoides sistémicos en estos pacientes no debe realizarse sin antes evaluar la correcta función de las glándulas suprarrenales mediante una prueba de estimulación de la ACTH, evitando así el riesgo de causar una insuficiencia suprarrenal aguda.
Además, debemos tener en cuenta que el uso de corticoides tópicos, especialmente en el caso de lesiones extensas o afectación de mucosas, puede ocasionar complicaciones debido a su absorción sistémica, aumentada en pacientes pediátricos.
Cura tópicaEn todos los pacientes con PA es importante mantener una higiene diaria y cuidadosa de la piel y mucosas, y es recomendable llevar un recuento aproximado de las lesiones. La higiene debe realizarse previamente a la aplicación del tratamiento tópico. Las ampollas pueden ser drenadas con una aguja o una hoja de bisturí estéril, incidiendo en la base de la lesión. La aplicación de una solución antiséptica como eosina al 2% o clorhexidina al 0,5% sobre las lesiones erosivas disminuye la posibilidad de sobreinfección. Si esto ocurre pueden utilizarse antibióticos tópicos como el ácido fusídico o la mupirocina. En caso de sospecha de infección cutánea profunda (celulitis) debe iniciarse antibioterapia sistémica.
Monitorización y manejo terapéuticoLa evolución clínica ha sido hasta hace poco el criterio sobre el que basar las decisiones terapéuticas a tomar en pacientes con PA. En general, en los casos en los que el tratamiento tópico es insuficiente o difícilmente aplicable, los corticoides sistémicos son el fármaco de elección (siempre que no estén contraindicados). En la mayoría de pacientes este tratamiento causa una mejoría clínica en pocas semanas, lo que nos permite reducir progresivamente su dosis y a menudo retirarlos en un plazo de 6 a 10 meses20. En aquellos pacientes con un PA resistente o dependiente de corticoides sistémicos a altas dosis estaría indicado añadir un fármaco adyuvante. Este tratamiento tiene como objetivo alcanzar una respuesta clínica aceptable y reducir la dosis de corticoides sistémicos. Sin embargo, no existe un consenso sobre cómo y cuándo variar el tratamiento de un paciente con PA.
Además de la valoración clínica, en nuestra experiencia, la determinación selectiva de anticuerpos circulantes frente al BP180 mediante ELISA es una herramienta útil para monitorizar a los pacientes, ya que se correlaciona con la actividad del PA y nos permite tomar decisiones terapéuticas más precisas.
Si esta técnica está disponible en el centro es recomendable obtener una medida inicial del título de anticuerpos anti-BP180 en el momento del diagnóstico, que podremos utilizar como referencia para el control del paciente.
En cuanto a la evaluación clínica, disponemos de sistemas de medida objetivos para monitorizar el estado clínico de los pacientes. De forma muy similar al Pemphigus Disease Area Index (PDAI), el Bullous Pemphigoid Disease Area Index (BPDAI) (fig. 5) mide separadamente la afectación cutánea y la afectación mucosa, y tiene en cuenta el tipo de lesión y la localización24.
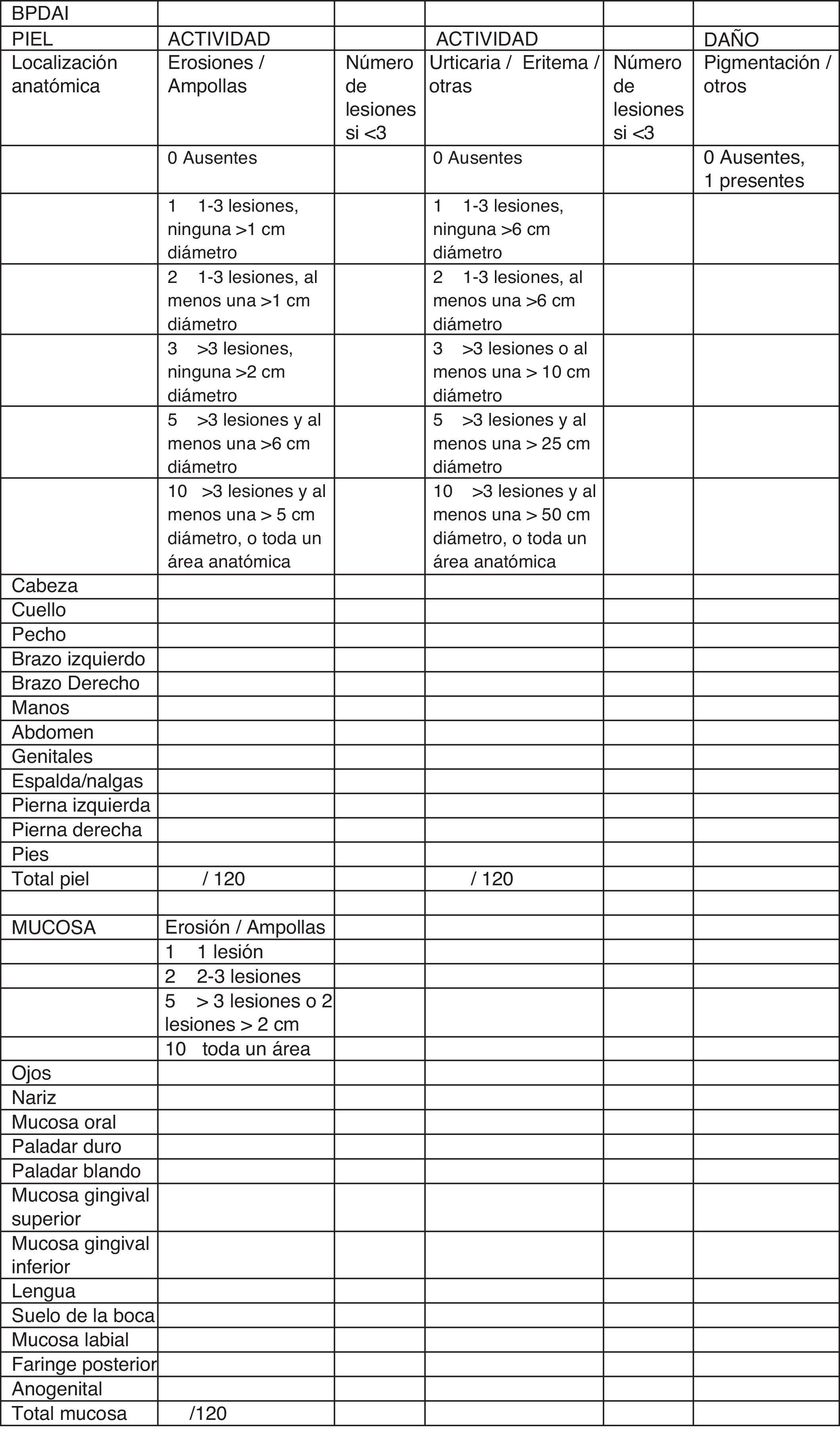
Bullous Pemphigoid Disease Area Index (BPDAI): escala de medida objetiva de la afectación cutáneo-mucosa en penfigoide ampolloso. Fuente: Murrell et al.24.
La Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) (fig. 6)25 también fue diseñada inicialmente para pénfigo vulgar, y así mismo separa las lesiones cutáneas de las mucosas y puntúa según el tipo de lesión y la extensión de la misma. El uso de estas escalas permite minimizar la subjetividad de la evaluación clínica, y es de gran utilidad para comparar resultados de forma fiable en el mismo paciente o con otros pacientes, y especialmente para comparar resultados de grupos distintos a nivel de estudios.

Autoimmune Bullous Skin disorder Intensity Score (ABSIS): escala de medida objetiva de la actividad clínica que puede utilizarse para penfigoide ampolloso. Fuente: Pfütze et al.25
Aún más subjetivo y difícil de valorar es el prurito. Se trata, sin embargo, de un síntoma que condiciona la calidad de vida del paciente y es por tanto importante evaluarlo. La escala visual analógica (EVA) (fig. 7) puntúa del 0 al 10 el grado de prurito presentado en las últimas horas, días y semanas, con una puntuación total de 0 a 30. En caso de que el paciente no sea capaz de responder a estas preguntas mediante la EVA, el prurito puede inferirse en función de las lesiones de rascado (fig. 7)24.

Escala visual analógica para la evaluación del prurito. Fuente: Murrell et al.24.
También respondiendo a la necesidad de comparar los resultados de los distintos estudios realizados sobre pacientes con PA, recientemente un grupo de expertos han consensuado un conjunto de definiciones útiles en la monitorización de la actividad de la enfermedad24. Los criterios de valoración aceptados por este grupo están recogidos en la tabla 124.
Criterios de valoración aceptados para penfigoide ampolloso
| Criterios de valoración iniciales | |
| Punto de partida | Día de inicio de tratamiento pautado para PA |
| Control de la actividad de la enfermedad | Momento en el que las lesiones estables inician la curación o el prurito comienza a ceder y no aparecen nuevas lesiones |
| Tiempo hasta el control de la actividad | Intervalo de tiempo que transcurre desde el punto de partida hasta el control de la actividad |
| Final de la fase de consolidación | No aparecen nuevas lesiones durante al menos 2 semanas, aproximadamente el 80% de las lesiones han curado y el prurito es mínimo |
| Lesiones transitorias | Nuevas lesiones que curan en una semana o prurito que dura una semana y se resuelve sin tratamiento |
| Lesiones persistentes | Nuevas lesiones que no curan en una semana o prurito que dura más de una semana con o sin tratamiento |
| Remisión completa durante la reducción de dosis de fármacos | Ausencia de lesiones con tratamiento a dosis superiores a la dosis mínima |
| Criterios de valoración finales | |
| Tratamiento con dosis mínimas | ≤0,1 mg/kg/d de prednisona (o equivalente) o 20 g/semana de propionato de clobetasol y/o terapia adyuvante mínima o de mantenimiento |
| Tratamiento con dosis mínimas adyuvante o de mantenimiento | Las siguientes dosis o dosis inferiores: metotrexato 5 mg/semana; azatioprina 0,7 mg/kg/d (con niveles TPMT normales); micofenolato de mofetilo 500 mg/d; ácido micofenólico 360 mg/d; o dapsona 50 mg/d |
| Remisión parcial en tratamiento con dosis mínimas | Presencia de nuevas lesiones transitorias que curan en una semana mientras el paciente recibe terapia mínima durante al menos 2 meses |
| Remisión completa en tratamiento con dosis mínimas | Ausencia de nuevas lesiones transitorias o estables o prurito mientras el paciente recibe terapia mínima durante al menos 2 meses |
| Remisión parcial sin tratamiento | Presencia de nuevas lesiones transitorias que curan en una semana sin tratamiento mientras el paciente no recibe medicación para el PA durante al menos 2 meses |
| Remisión total sin tratamiento | Ausencia de nuevas lesiones o lesiones estables o prurito mientras el paciente está sin medicación para el PA durante al menos 2 meses |
| Nueva actividad leve de la enfermedad | <3 Lesiones al mes que no curan en una semana, o extensión de lesiones estables o prurito una vez/semana, pero no diario en un paciente que ha alcanzado el control de la enfermedad; estas lesiones deben curar en 2 semanas |
| Recidiva/brote | Aparición de ≥3 nuevas lesiones al mes o al menos una lesión grande (>10cm diámetro) que no cura en una semana, o extensión de lesiones estables o prurito diario en un paciente que ha alcanzado el control de la enfermedad |
| Fallo de control inicial con tratamiento | Aparición de nuevas lesiones estables o extensión de lesiones antiguas o fallo en la curación de lesiones estables o prurito continuo a pesar de: propionato de clobetasol 40g/d durante 4 semanas. Prednisona 0,75mg/kg/d o equivalente durante al menos 3 semanas con o sin tratamiento adyuvante. Tetraciclinas a dosis completas durante 4 semanas. Dapsona 1,5 mg/kg/d durante 4 semanas. Metotrexato 15mg/sem (si >60kg peso y sin fallo renal) durante 4 semanas. Azatioprina 2,5mg/kg/d durante 4 semanas (con niveles TPMT normales). Micofenolato de mofetilo 40mg/kg/d (con función renal normal) durante 4 semanas |
Fuente: Murrell et al.24
PA: penfigoide ampolloso; TPMT: tiopuril metil transferasa.
Los corticoides tópicos o sistémicos son en general el tratamiento de primera elección en un paciente con PA. El uso de corticoides tópicos de alta potencia (tabla 2) está recomendado especialmente en pacientes con clínica leve o moderada, en los que puede ser el único tratamiento necesario, evitando las complicaciones de los fármacos sistémicos. Además se utilizan frecuentemente como tratamiento complementario adyuvante.
Corticoides tópicos
| Monitorización previa al Tratamiento | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| No precisa | No precisa | Propionato de clobetasol 0,05%10-40g/d | No se recomienda su uso sobre lesiones cutáneas infectadas ya sea por virus, hongos o bacteriasHipersensibilidad al principio activoRosácea, acné vulgar y dermatitis perioral | No observadas | Dilatación de los vasos sanguíneos superficiales, atrofia cutánea local, estrías, hipertricosis, sobreinfección localRaros: hipersensibilidad al fármacoPor absorción sistémica: síndrome de Cushing, insuficiencia suprarrenal, hiperglucemia, cataratas, glaucoma, reactivación tuberculosis |
Su eficacia como tratamiento único ha sido descrita en numerosos casos y series cortas de pacientes. En un estudio reciente con 96 pacientes de PA el 62% consiguió controlarse solo con corticoides tópicos (dosis media de 30g/d de propionato de clobetasol al 0,05%)26. En un estudio aleatorizado se demostró que eran más eficaces que los corticoides sistémicos, y que tenían además un mejor perfil de seguridad22. El único inconveniente que tenía este estudio es que se utilizó una pauta de administración poco convencional, que consistía en aplicar 40g de propionato de clobetasol diarios por toda la superficie cutánea (incluyendo piel sana). Esta pauta ha sido bastante criticada, dado que es complicada de realizar en pacientes ancianos, y se desconoce cuál es la cantidad real de corticoides que se absorben (de hecho nosotros hemos observado cuadros de síndrome de Cushing con esta pauta de forma similar a lo que sucede con los corticoides sistémicos). En un estudio posterior del mismo grupo se describe la misma eficacia de dosis inferiores de clobetasol tópico, pero con menos efectos secundarios27. Por otro lado, debemos recordar que a menudo los pacientes con PA pueden no ser lo suficientemente autónomos como para aplicarse la medicación sin ayuda. Esto dificulta, sin duda, la adherencia a este tipo de tratamiento, lo que puede disminuir notablemente su eficacia.
Corticoides sistémicosLa eficacia de los corticoides sistémicos (tabla 3) ha sido demostrada por diversos estudios y está establecida por una amplia experiencia clínica. En la mayoría de los casos se consideran el tratamiento de elección si la terapia tópica no es suficiente o no puede ser aplicada y siempre que no estén contraindicados. Los fármacos más empleados en este grupo son la prednisona y la prednisolona.
Corticoides sistémicos
| Monitorización previa al Tratamiento | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma, bioquímica básica, perfil hepático y renalSerologías víricas: VHC, VHB y VIHPresión arterialRadiografía de tóraxTest de Mantoux | Hemograma y bioquímica básica | Dosis inicial recomendada:0,5-0,75mg/kg/dSi rebrote: aumentar 10-20 mg/d cada 2 o 3 semanas hasta control clínico | Relativas: diabetes mellitus, osteoporosis grave, sangrado digestivo, pseudotumor cerebral, aplasia, psicosis, miopatía esteroidea y glaucomaAbsolutas: herpes simple ocular, TBC activa | AINE (úlcera péptica), anfotericina B (riesgo de hipocalemia), anticoagulantes derivados de la cumarina, heparina, estreptoquinasa o uroquinasa (disminuye efecto anticoagulante), antidepresivos tricíclicos, anticonceptivos orales (incrementan la vida media de los corticoides), glucósidos digitálicos (aumentan el riesgo de arritmias) | Aumento de peso, hiperglucemia, osteoporosis, supresión suprarrenal, úlcera péptica, alteraciones del humor y del sueño, síndrome de Cushing, cataratas, miopatía proximal y aumento del riesgo de infección |
AINE: antiinflamatorios no esteroideos; TBC: tuberculosis; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
La dosis recomendada está entre 0,5 y 0,75mg/kg/d, dado que dosis superiores no han demostrado aumentar la eficacia del tratamiento, y sí que comportan un mayor riesgo de efectos secundarios28. Es necesario recordar que a estas dosis debe realizarse tratamiento preventivo de úlcera péptica gastroduodenal con inhibidores de la bomba de protones, e instaurar desde el inicio tratamiento con suplementos de calcio, vitamina D y bifosfonatos para prevenir la osteoporosis secundaria a los esteroides.
En general, observaremos una respuesta favorable a los corticoides sistémicos ya en las primeras semanas de tratamiento, lo que nos permite entonces reducir la dosis. A menudo conseguimos una remisión completa en un plazo de 6-10 meses, pudiendo suspender totalmente este tratamiento21.
Tetraciclinas, eritromicina y nicotinamidaCuando la respuesta a la corticoterapia es incompleta o aparecen efectos secundarios, fármacos no inmunosupresores como tetraciclinas, eritromicina, nicotinamida o sulfona pueden ser utilizados como tratamiento adyuvante, especialmente en pacientes con una clínica leve o moderada. Las propiedades antiinflamatorias de las tetraciclinas y la nicotinamida (tabla 4) permiten su utilización en procesos que tienen un mecanismo inmunológico en su patogenia como el PA. Además, la ausencia de efectos secundarios importantes facilita su empleo en personas mayores y en niños. En el caso de las tetraciclinas se ha observado capacidad de inhibir la quimiotaxis de neutrófilos y eosinófilos, además de inhibir la acción de ciertas metaloproteasas. Suelen asociarse a nicotinamida, que actúa sobre diferentes puntos de la respuesta inflamatoria.
Tetraciclinas/eritromicina/nicotinamida
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma completo y bioquímica básica, perfil hepático y renal | No precisa | Tetraciclina: 0,5-2g/dEritromicina: 1-3g/dDoxiciclina: 200-300mg/d Minociclina: 100-200mg/dNicotinamida: 500mg-2,5g/d | Tetraciclina: insuficiencia renal y niños (<12 años)Doxiciclina y minociclina: insuficiencia hepáticaEritromicina: contraindicación relativa en insuficiencia hepática | Las tetraciclinas reducen el efecto de los anticonceptivos y potencian el de los anticoagulantes oralesLos antiácidos y las sales de aluminio, calcio, hierro, magnesio y cinc reducen la absorción de las tetraciclinas | Frecuentes: alteraciones gastrointestinales (colitis asociada a los antibióticos de forma ocasional), vértigos, fototoxia, disfagia e irritación esofágicaRaros: hepatotoxicidad, pancreatitis, trastornos hematológicos y reacciones de hipersensibilidad |
La mayoría de los datos sobre la eficacia de estos tratamientos provienen de casos únicos aislados. Sin embargo, disponemos de un estudio aleatorizado sobre una serie pequeña de pacientes que no encontró diferencias significativas en cuanto a respuesta entre pacientes con PA tratados con tetraciclina 2g/d combinada con niacinamida 2g/d y pacientes tratados únicamente con prednisona 40-80g/d29.
SulfonaEl mecanismo de acción de la sulfona (tabla 5) no ha sido totalmente descifrado, aunque también parece presentar un efecto antiinflamatorio, dado que es capaz de inhibir la adhesión de los neutrófilos al endotelio de los vasos, la quimiotaxis, la producción de lipooxigenasa y la acción de la mieloperoxidasa de los neutrófilos y eosinófilos. No existen estudios aleatorizados sobre el uso de sulfona, ya sea como tratamiento único o adyuvante en el tratamiento del PA. No obstante, varios estudios retrospectivos han observado una respuesta de entre el 15 y el 45% a este fármaco30–33. Un inconveniente importante de la sulfona es la presencia casi constante de anemia asociada a dosis terapéuticas del fármaco. Pueden ser normales descensos de 1-2g/dl de las cifras de hemoglobina, en ocasiones muy mal tolerados en pacientes ancianos (nosotros hemos observado la aparición de cuadros de angor, o de insuficiencia cardíaca en algunos pacientes).
Sulfonaa
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma y reticulocitos, perfil renal con electrolitos, perfil hepático, G6PDH | Hemograma, reticulocitos, metahemoglobina, perfil renal y hepático cada 2-4 semanas hasta 3 meses después | Dosis inicial recomendada:50mg/d Mantenimiento:100mg/día (dosis máxima 200mg/d)Niños:0,5 a 2mg/kg/d | Alergia a sulfonamidas, anemia severa, déficit de G6PDH y porfirias agudas | Quinidina, digoxina, ciclosporina, eritromicina, neomicina, tetraciclina, amiodarona, diltiazem, propafenona, quinidina y verapamilo | Frecuentes: hemólisis y metahemoglobulinemia (dosis dependiente)Poco frecuentes: síndrome de hipersensibilidad a fármacos (tipo DRESS), erupción morbiliforme que puede progresar a eritrodermia, adenopatías, hepatitis, neutropenia, neuropatía periférica, psicosis |
DRESS: drug reaction with eosinophilia and systemic symptoms; G6PDH: glucosa 6 fosfato deshidrogenasa.
En los casos de PA moderado o grave en los que la corticoterapia a dosis altas no consigue controlar la actividad de la enfermedad, o no es posible mantener la dosis eficaz debido a los efectos secundarios, estaría indicada la administración de fármacos inmunosupresores.
MetotrexatoEn nuestra experiencia el metotrexato (MTX) (tabla 6) es uno de los fármacos inmunosupresores de primera línea en el PA. Se trata de un antagonista del ácido fólico que se utiliza como ahorrador de corticoides a dosis bajas en numerosas enfermedades. Para los dermatólogos presenta la ventaja de tratarse de un fármaco inmunosupresor ampliamente conocido dado su uso frecuente en el tratamiento de la psoriasis desde hace décadas, con un perfil de seguridad muy bueno.
Metotrexatoa
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma, bioquímica básica sangre y orina, perfil hepático y renalSerologías VHB, VHC, VIHFerritina e índice saturación transferrina, anticuerpos antimitocondrialesPropéptido amino-terminal del procolágeno tipo iii (PPIII)Ecografía hepática si hepatopatía previaTest embarazoRadiografía de tóraxTest de Mantoux | Hemogramab, función renal, electrolitos, función hepática (semanal tras aumento de dosis, quincenal por un mes, mensual por 3 meses, luego cada 3-4 meses)PPIII cada 3-6 mesesVigilar aumento de transaminasas y de PPIIIc | 2,5 a 15mg por semanadPor encima de 15mg la biodisponibilidad disminuye en la presentación oralPuede ser oral, subcutáneo o intramuscular | Embarazoe, lactancia, citopenias, enolismo, insuficiencia hepática o renal severa (filtrado glomerular <10 ml/min/m2)Precaución si deshidratación, hepatopatía previa o infección activa | Antagonistas del ácido fólico (sulfamidas, trimetoprim, fenitoína), AINE, azatioprina, ciclosporina, probenecid, radioterapia y retinoides orales. Desaconsejadas vacunas vivas durante el tratamiento | Frecuentes: alteraciones gastrointestinales, fatiga y malestar general (son dosis dependientes y aumentan tras la toma del fármaco, mejor tomar en día no laborable)Poco frecuentes: macrocitosis, mielosupresión, hepatotoxicidad aguda/crónica, neuropatía, mucositis y osteopatía tibialBajo potencial oncogénicoRiesgo de reactivación de tuberculosis |
AINE: antiinflamatorios no esteroideos; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
Se recomienda añadir ácido fólico (diario), o folínico (semanal) para mejorar la tolerancia gastorintestinal, y disminuir la toxicidad.
Aumento del volumen corpuscular medio (VCM): administrar suplementos de ácido fólico. Si persiste VCM elevado (>106fl): suspender metotrexato.
Aumento de transaminasas: Si aumento <3 veces su límite superior: reducir dosis. Si la alteración persiste suspender el fármaco. Si aumento >3 veces su límite superior: suspender metotrexato. Aumento persistente PPIII: suspender fármaco.
Las dosis utilizadas para el tratamiento del PA son en general bajas, entre 2,5 (en pacientes ancianos con función renal deteriorada) y 10mg semanales. En algunos pacientes puede llegarse a dosis superiores, pero como máximo suelen ser de 15mg/semana. Es muy importante indicar bien al paciente (o a sus cuidadores, dado que a menudo son pacientes dependientes que se hallan ingresados en residencias) que el MTX se debe administrar en una dosis única semanal. Se han dado casos (en pacientes con psoriasis) en los que se cambió la dosificación a dosis diarias por desconocimiento del fármaco por parte del personal sanitario a cargo del paciente, provocando el fallecimiento por los efectos tóxicos del MTX.
La evidencia sobre la eficacia de este tratamiento en el PA recae sobre numerosos casos y series de casos en los que se ha utilizado, ya sea como fármaco ahorrador de corticoides sistémicos en pacientes con enfermedad moderada-grave, en monoterapia o en combinación con corticoides tópicos en pacientes con menor gravedad34,35. Un estudio prospectivo con 11 pacientes a los que se administró MTX y corticoides tópicos, consiguiendo una rápida respuesta en todos ellos, recomienda iniciar el fármaco a 5mg/semana y aumentar 2,5mg cada semana (máximo 12,5mg/semana) hasta conseguir la remisión clínica34.
Otros fármacos inmunosupresores más potentes como la azatioprina (AZA), el micofenolato (MF) o la ciclofosfamida (CF) estarían indicados en casos de PA grave o refractario.
AzatioprinaLa AZA (tabla 7) es un derivado imidazólico de la 6-mercaptopurina, activo por vía oral y parenteral, con propiedades antiinflamatorias e inmunosupresoras. Hasta el momento no se ha demostrado una diferencia significativa en cuanto a la respuesta en pacientes con PA tratados con AZA como adyuvante y pacientes tratados únicamente con corticoides sistémicos36,37. Sin embargo, existe amplia experiencia sobre su uso como fármaco ahorrador de corticoides, siendo en nuestro medio el fármaco más frecuentemente pautado con este fin.
Azatioprina
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Niveles TPMTa, hemograma, función renal, función hepática, serología virus varicela zóster. Test embarazo si procede | Hemograma, función renal y función hepática 2 semanas tras inicio o cambio de dosis; al mes y cada 2-3 meses | Dosis inicial recomendada: 50mg/12hDosis según TPMTa:<5U/ml: contraindicada5-14U/ml: 0,5 mg/kg/d14-18U/ml: 1,5 mg/kg/d18-26U/ml: 2,5 mg/kg/d> 26U/ml: 3 mg/kg/dAjustar en insuficiencia renal y hepática controlando VCM | Embarazo y lactancia, alergia a AZA o 6-mercaptopurina, neoplasia concurrenteTPMT<5Precaución: insuficiencia renal y hepática o antecedentes de neoplasia en remisión | Alopurinol, anticoagulantes orales, cimetidina, cotrimoxazol, IECA, indometacina, penicilamina, sulfasalazinaNo administrar vacunas vivas durante su uso | Frecuentes: molestias gastrointestinales y aftas oralesPoco frecuentes: macrocitosis, mielosupresión, hepatotoxicidad, reacciones de hipersensibilidad (idiosincrática), pancreatitis, alopecia difusa (reversible). Bajo potencial oncogénico |
IECA: inhibidores de la enzima convertidora de angiotensina; VCM: volumen corpuscular medio.
En nuestro medio hay un 0,3% de la población con actividad baja de TPMT y un 11,9% con actividad intermedia. Estos pacientes pueden presentar graves efectos secundarios con la toma de azatioprina (AZA) como hepatitis colestásica, mielosupresión severa, pancreatitis o toxicidad gástrica. Los pacientes con actividad elevada de tiopuril metil transferasa (TPMT) (>80% de la población) pueden precisar dosis elevadas del fármaco para que sea eficaz, y son los que suelen tener mayor riesgo de hepatoxicidad.
El efecto terapéutico se obtiene normalmente entre las 6 y las 10 semanas de tratamiento. Previamente al inicio del tratamiento con AZA deberíamos conocer la actividad de la tiopurina metil transferasa (TPMT) del paciente. Esta enzima metaboliza el fármaco y el conocimiento de su actividad permite ajustar su dosis y evitar la toxicidad hematológica, aunque no aporta información sobre la toxicidad hepática. La actividad de la TPMT depende de polimorfismos genéticos de 3 alelos (TPMT*2, TPMT*3A y TPMT*3C), que están involucrados en el 95% de los casos de actividad enzimática baja o intermedia. La presencia de estos 3 alelos es predictiva del fenotipo: los pacientes heterocigotos (un alelo salvaje y una de las variantes alélicas) tendrán una actividad intermedia de la enzima TPMT, mientras que los homocigotos (los 2 alelos corresponden a las variantes alélicas, esto sucede en 1 de cada 300 individuos de la población general) son totalmente deficientes en la actividad de la enzima. La determinación de la actividad de la TPMT se puede realizar mediante 2 métodos: determinar la actividad intraeritrocitaria de la enzima (que es menos precisa) o determinar los alelos genéticos mediante PCR.
MicofenolatoEl micofenolato (tabla 8) es un antagonista de la síntesis de ADN-nucleótidos. Existen 2 fármacos diferentes que se emplean en la clínica. El micofenolato de mofetilo (MFM) es un profármaco del ácido micofenólico que se emplea desde 1997 en la prevención del rechazo en trasplante renal, y actualmente está indicado (según ficha técnica) además para trasplante hepático y cardiaco. El micofenolato sódico con cubierta entérica (MFS-CE) ha sido diseñado con la intención de disminuir la intolerancia gastrointestinal asociada con mucha frecuencia al MFM. Esta cubierta consigue retrasar la liberación del fármaco evitando su contacto con el medio ácido del estómago. Actualmente está aprobado por la FDA para trasplante renal.
Micofenolato de mofetil (MFM) y micofenolato sódico con cubierta entérica (MFS-CE)
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma y bioquímica general, perfil hepáticoTest embarazo si procede | Hemograma y bioquímica mensualmente hasta el 3.er mes y posteriormente cada 2-3 mesesSuspender si neutrófilos <1300cél/ml | MFM: 1g/12hMFS-CE:720mg/12hAjustar en pacientes con insuficiencia renal | Embarazo, lactancia y alergia al fármacoPrecaución en úlcera péptica o gastritis erosiva | Aciclovir, antiácidos con aluminio o magnesio, azatioprina, colestiramina, ganciclovir, probenecid. No administrar vacunas vivas | Frecuentes: molestias gástricasPoco frecuentes: astenia, síntomas genitourinarios, linfopenia y mielosupresión, elevación de transaminasas, mayor riesgo de infecciones y de neoplasias |
Disponemos aún de pocos estudios sobre el uso de MFM o de MFS-CE en el tratamiento del PA. En general, se utiliza de modo similar a la AZA, con el objetivo de ahorrar corticoides sistémicos, aunque también se ha usado en monoterapia con buena respuesta38. Presenta una eficacia similar a la AZA y un perfil de seguridad mejor, pero su precio es bastante más elevado. Un estudio reciente no encontró diferencias significativas en cuanto a eficacia entre pacientes con PA tratados con corticoides y AZA y pacientes tratados con corticoides y MFM39.
CiclofosfamidaLa toxicidad de la CF (tabla 9), especialmente su efecto sobre la fertilidad, la mielosupresión y el aumento de riesgo de neoplasias, limita de forma importante su uso a situaciones en las que otros fármacos están contraindicados o son ineficaces. Se trata de uno de los inmunosupresores más potentes, tanto de la inmunidad celular como de la humoral, pero también de los más eficaces. Es un agente alquilante, de uso oral o intravenoso y que se metaboliza por el citocromo P450. Aunque no disponemos de estudios aleatorizados sobre su utilidad en PA, existen publicaciones de casos o series cortas de pacientes en los que la CF se utiliza como fármaco adyuvante, ahorrador de corticoides, considerándose un fármaco potente pero también bastante más tóxico que otros inmunosupresores40,41. En nuestra experiencia personal puede ser una opción muy válida como segunda línea, utilizado a dosis bajas de 50 a 100mg/día, particularmente en pacientes ancianos en los que quizás no preocupan tanto los efectos secundarios a largo plazo18 (Gual et al. Manuscrito sometido a publicación).
Ciclofosfamida
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma, perfil hepático y renal (con electrolitos), sedimento urinarioTest embarazo si procede | Hemograma, perfil hepático y renal y sedimento urinario cada 2 semanas tras inicio y cambio de dosis, después mensualmente | Vía oral: 1-3mg/kg/d (máx. 200mg/día) Debe ajustarse en insuficiencia renal y hepáticaEndovenosa: 500-1.000 mg/m2 según tolerancia | Embarazo y lactancia, hipersensibilidad al fármaco, cistitis hemorrágica, infección urinaria, infección sistémica aguda, medicación o radioterapia con toxicidad urotelial y mielosupresión | Alopurinol, anticoagulantes orales, antidiabéticos orales, digoxina, doxorrubicina, insulina, quinolonas, suxametonio, tiacidas y vacunas vivas | Náuseas, vómitos, anorexia, azoospermia e insuficiencia ovárica (irreversible a dosis de 4-5g totales), SIADH, mielotoxicidad, alopecia (efluvio anágeno), cistitis hemorrágica (suspender fármaco), toxicidad cardiaca y pulmonar y aumento del riesgo de infecciones y neoplasias (especialmente vejiga) |
SIADH: síndrome de secreción inadecuada de ADH.
En los últimos años nuevas terapias no inmunosupresoras, con distintos mecanismos de acción como las inmunoglobulinas endovenosas y el rituximab, han supuesto una alternativa de tercera línea, especialmente en casos de PA grave o refractario (fig. 8).
Inmunoglobulinas intravenosas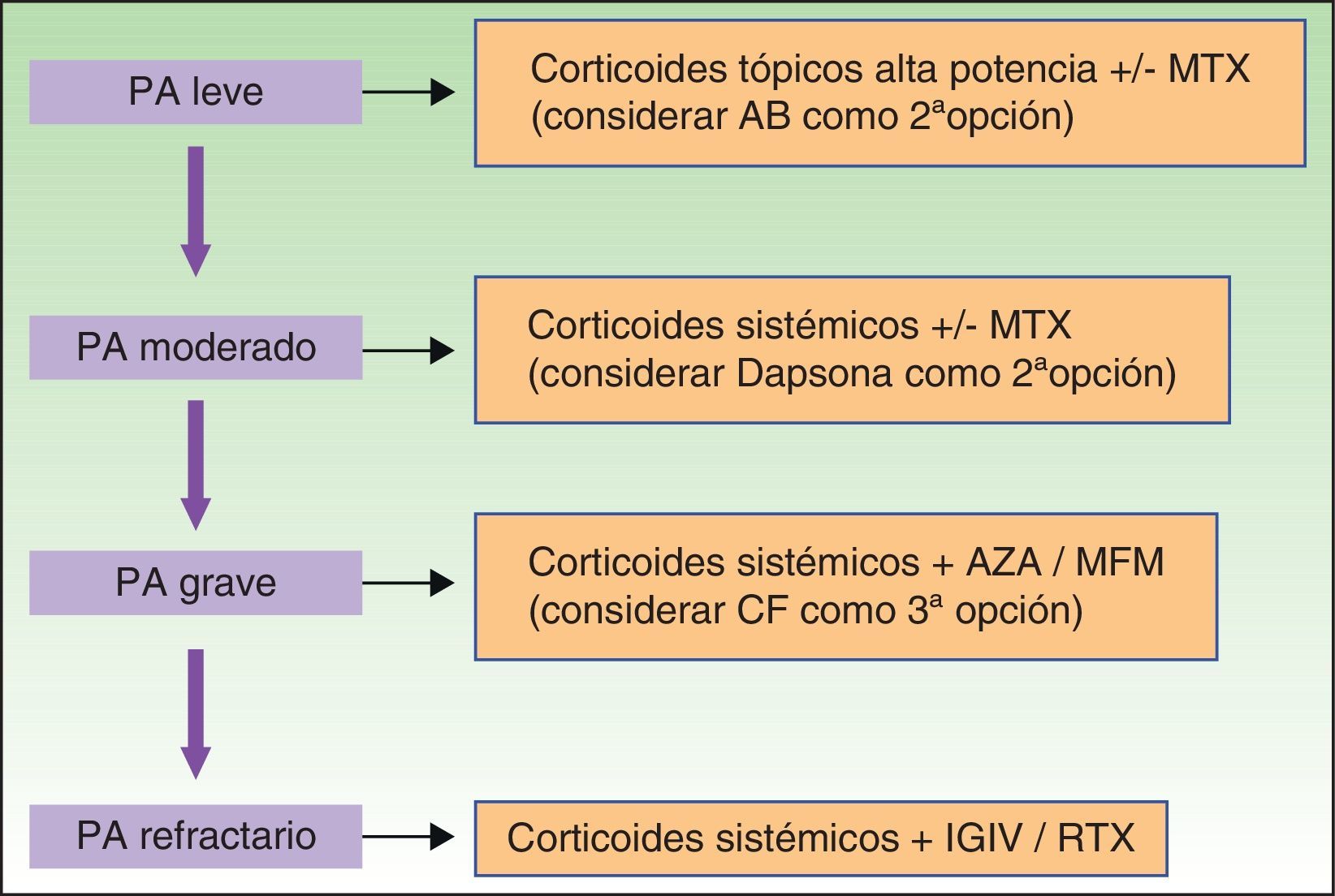
Las inmunoglobulinas intravenosas (IGIV) (tabla 10) se obtienen a partir del plasma de numerosos donantes. Aproximadamente el 90% de las inmunoglobulinas presentes en cada preparado son IgG y el resto IgA e IgM. Su mecanismo de acción es complejo, y no ha sido completamente aclarado. En diversas enfermedades autoinmunes la administración de IGIV se ha relacionado con un descenso claro y rápido de los niveles de anticuerpos patogénicos. Esto ha sido demostrado también en el caso de los anticuerpos anti-membrana basal en el PA, y también por estudios experimentales en animales42.
Inmunoglobulinas intravenosasa
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma, perfil hepático y renal, glucemia, dosificación de inmunoglobulinas (el déficit de IgA aumenta el riesgo de anafilaxia)Serologías VHB, VHC y VIH, crioglobulinas, grupo sanguíneo, presión arterial | Hemograma, perfil hepático y renal, glucemia, presión arterial | 2g/kg repartidos durante 3 a 5 díasRepetir cada 3 semanasNormalmente 2-4 ciclosSe recomienda utilizar siempre el mismo tipo en cada paciente | Déficit absoluto de IgA, fallo renal severo e hipersensibilidad grave a IGIVPrecaución en ancianos, artritis reumatoide, cardiopatía, crioglobulinemia, hipercoagulabilidad, lupus eritematoso sistémico, migraña y nefropatíaSi riesgo alto de tromboembolismo: administrar 500ml de suero fisiológico antes y después de la perfusión y AAS 100mg o heparina cálcica 1.000UI durante 3 días | No mezclar IGIV con otras soluciones endovenosas en la misma víaEvitar administración con fármacos neurotóxicos y vacunas vivas | Frecuentes: reacción infusional (temblor, taquicardia, HTA, lumbalgia, febrícula, mialgias, náuseas y vómitos, cefalea, flushing, dolor torácico, disnea)Poco frecuentes: meningitis aséptica autolimitada, descompensación cardiopatía/nefropatía, HTA, fenómenos tromboembólicos, rash, infecciones, empeoramiento de migraña, anemia hemolítica y anafilaxia |
AAS: ácido acetil salicílico; IGIV: inmunoglobulinas endovenosas; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
La infusión debe ser lenta (aproximadamente 6h, empezando por 15ml/min durante 15min) y es conveniente premedicar con desclorfeniramina y paracetamol al paciente una hora antes. Si durante la infusión el paciente presenta sensación de opresión, rubeosis o hipotensión debe reducirse la velocidad de perfusión. Si los síntomas no mejoran puede requerirse desclorfeniramina endovenosa, corticoides e incluso adrenalina. Si aparece una reacción severa debe suspenderse la perfusión del fármaco.
Aunque el tratamiento de PA no se incluye entre las indicaciones del tratamiento con IGIV aprobadas por la FDA, en los últimos años se han descrito numerosos casos de PA tratados eficazmente con este fármaco. En la mayoría de ellos, incluido un estudio prospectivo con 15 pacientes, se trata de casos de PA refractarios a otras terapias y que respondieron adecuadamente a las IGIV43. Se ha observado que el uso de fármacos inmunosupresores junto a las IGIV disminuye el riesgo de efecto rebote tras el descenso rápido de anticuerpos patógenos provocado por las IGIV.
Las IGIV son un tratamiento caro, de acción rápida y no inmunosupresor. Su administración requiere disponibilidad de hospital de día o ingreso hospitalario. Dado su elevado coste solo estarían indicadas en pacientes que no responden o presentan efectos secundarios a otros tratamientos de segunda línea, o si se necesita una respuesta rápida y no se quiere inmunosuprimir más al paciente.
RituximabEl rituximab (RTX) (tabla 11) es un anticuerpo monoclonal, quimérico, humanizado cuyas células diana expresan CD-20. Esto incluye células pre-B y linfocitos B maduros, pero no células madre ni plasmáticas. Este fármaco fue inicialmente desarrollado para el control de procesos linfoproliferativos de células B; sin embargo, su eficacia en el control de diversas enfermedades autoinmunes refractarias a otros tratamientos, ya sea solo o en combinación con IGIV, está siendo demostrada para un número cada vez mayor de enfermedades en los últimos años.
Rituximab
| Monitorización previa | Monitorización durante el tratamiento | Dosis utilizadas | Contraindicaciones | Interacciones | Efectos secundarios |
| Hemograma y recuento de linfocitos B, serologías: VHB, VHC, VIH. PPD y radiografía de tóraxValorar vacuna antineumocócica | Hemograma y recuento linfocitos B | Dosis más usada: 375mg/m2 por semana, durante 4 semanasValorar administración única si recaída | Hepatitis B o C activa, hipersensibilidad a proteínas de ratón (murinas), infección activa no controlada, infección por VIH con linfocitos T CD4<250cél/μl e insuficiencia cardíaca graveEmbarazo y lactancia (se recomienda anticoncepción hasta un año después de acabar el tratamiento) | Evitar uso de vacunas con virus atenuados y fármacos anti-TNF (hasta 16 semanas después de última dosis) | Frecuente: reacción infusionalPoco frecuentes: hipotensión, erupción urticariforme/angioedema, broncoespasmo, trombopenia (habitualmente tras primera infusión y reversible), aumento del riesgo de infecciones, necrólisis epidérmica tóxica, leucoencefalopatía multifocal progresiva, arritmias e insuficiencia cardíaca |
Debe administrarse de forma lenta (5-6 horas la 1.a infusión) y se recomienda premedicación con antihistamínicos y corticoides endovenosos (generalmente solo antes de la 1.a infusión). Algunos autores recomiendan una infusión de IGIV 400mg/kg en dosis única el día anterior a la infusión de rituximab como profilaxis de infecciones.
PPD: derivado proteico purificado (tuberculina); VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
El efecto del RTX tarda aproximadamente un mes y se mantiene durante 9-12 meses (hasta que se recuperan los linfocitos B). Este fármaco ha demostrado su eficacia en enfermedades ampollosas autoinmunes con mayor morbimortalidad como el pénfigo vulgar o foliáceo. Por otro lado, la implicación de los linfocitos B en la fisiopatología del PA justifica su valoración en caso de enfermedad refractaria. Sin embargo, la experiencia en cuanto al tratamiento del PA con RTX es aún escasa (casos aislados) y no se dispone de estudios aleatorizados en cuanto al manejo de este fármaco en estos pacientes (dosificación, monitorización).
En un estudio retrospectivo reciente en el que 5 pacientes con PA refractario a diversos fármacos se trataron con RTX, se observó una respuesta completa en 3 de los pacientes y parcial en un paciente. El otro paciente, cardiópata, murió pocos días después del inicio del tratamiento de forma inesperada44.
El RTX debe ser administrado en régimen de ingreso hospitalario u hospital de día. Presenta una toxicidad y una mortalidad asociada no despreciable, especialmente por el riesgo de infecciones graves, oportunistas o no. Por este motivo es un fármaco a tener en cuenta en pacientes con enfermedad grave que no responde al tratamiento habitual45.
PlasmaféresisLa plasmaféresis es una técnica costosa, no disponible en la mayoría de centros y por tanto poco utilizada en nuestro medio, incluso para el tratamiento de las enfermedades ampollosas autoinmunes de mayor morbilidad. Consiste en el recambio de un volumen importante de sangre del paciente (2 a 5l) por sucedáneos de plasma.
Dos estudios aleatorizados han comparado los efectos de esta técnica unida a los corticoides sistémicos versus corticoterapia en monoterapia36 o con AZA46. No se apreciaron diferencias entre la plasmaféresis y la AZA como tratamientos adyuvantes. Sin embargo, en uno de los estudios46 observaron una reducción de la dosis requerida de corticoides en los pacientes tratados con plasmaféresis.
Este tratamiento aumenta el riesgo de trombosis, infecciones y alteraciones electrolíticas. Además, requiere la realización de una fístula arterio-venosa, con los efectos secundarios que esta conlleva. Suele administrarse combinada con corticoides sistémicos o fármacos inmunosupresores, que ayudan a disminuir el efecto rebote producido por la activación de linfocitos autorreactivos tras el recambio plasmático.
La plasmaféresis es por tanto un tratamiento excepcional y raramente usado para el PA. Estaría reservado para casos agudos y graves con contraindicaciones para otros tratamientos y con una alta concentración de anticuerpos circulantes.
Conclusiones basadas en la evidenciaLa última revisión sobre el manejo del PA publicada en The Cochrane Library en 201047 incluye 10 estudios aleatorizados sobre el tratamiento del PA. Extrae como principales conclusiones las siguientes:
- 1.
Los corticoides tópicos de alta potencia son fármacos eficaces y presumiblemente seguros.
- 2.
Las dosis iniciales superiores a 0,75mg/kg/d de prednisolona no han demostrado mejorar la respuesta clínica.
- 3.
No se ha podido demostrar un aumento en la eficacia con el uso de plasmaféresis o AZA como adyuvantes a los corticoides sistémicos.
- 4.
No se ha demostrado diferencias entre la eficacia de la AZA y el micofenolato de mofetilo como adyuvantes a los corticoides sistémicos.
- 5.
La combinación de tetraciclinas y nicotinamida parece ser eficaz, aunque se necesitan estudios para confirmarlo.
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.


