





INTRODUCCIÓN
La papulosis linfomatoide (PL) fue definida inicialmente por Macaulay (1) en 1968 como «una erupción papulosa, rítmica, paradójica, clínicamente benigna, pero histológicamente maligna».
Se trata de una dermatitis crónica de curso clínico benigno, recurrente, con remisiones espontáneas, que histológicamente presenta un denso infiltrado linfoide de aspecto maligno, por lo que ha sido considerada como un pseudolinfoma T (2). Pero a pesar de presentar una evolución generalmente benigna, recientemente en la clasificación de los linfomas cutáneos de la EORTC (3) aparece incluida dentro de los linfomas T cutáneos de bajo grado. Además en el 10-20% de los pacientes la PL es precedida, coincidente o seguida de otro tipo de linfoma.
Las lesiones de la PL ocurren generalmente en la edad adulta (adultos jóvenes), siendo excepcional su incidencia en la niñez (4). De los casos descritos en pacientes menores de 20 años, más de la mitad aparecen en la primera década de la vida (5), siendo el comienzo más precoz descrito hasta ahora a los 8 meses de edad (6).
Presentamos el caso de una niña de 23 meses de edad con lesiones de características clinicopatológicas típicas de papulosis linfomatoide tipo A.
DESCRIPCIÓN DEL CASO
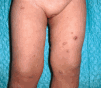
Una niña de 23 meses de edad, sin antecedentes personales de interés, acudió a nuestra consulta en febrero de 2000 por presentar una erupción cutánea de 15 días de evolución compuesta por lesiones asintomáticas, inicialmente localizadas en muslo izquierdo con posterior generalización de forma eruptiva por tronco y extremidades. No presentaba fiebre, malestar general, ni otra sintomatología asociada. A la exploración física presentaba múltiples (más de 50) lesiones papulonodulares infiltradas, con superficie cupuliforme de coloración rojizo-marrón y tamaño variable, hasta aproximadamente 1 cm de diámetro. La distribución era extensa, de predominio en tronco y miembros (Fig. 1), pero también estaban localizadas a nivel facial, cervical y ambos glúteos. No se palpaban adenopatías ni organomegalias.
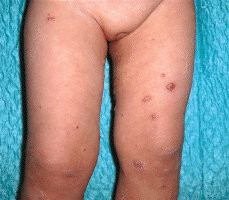
FIG. 1.--Lesiones cutáneas papulosas, papulonecróticas y/o nodulares en diferente estadio evolutivo, presentes simultáneamente en región proximal de miembros inferiores.
Estas lesiones crecieron rápidamente durante algunos días, desarrollando centros necróticos ulcerados y remitieron lentamente de forma espontánea, entre 3 y 6 semanas, dejando una fina cicatriz atrófica circular.
El estudio histopatológico de una de las lesiones cutáneas (Fig. 2) evidenciaba una epidermis hiperplásica, con discreta espongiosis e hiperqueratosis paraqueratósica, con abundantes polimorfonucleares en la capa córnea; bajo ella se observaba un denso infiltrado celular superficial y profundo constituido por linfocitos, células atípicas de aspecto linfoide con núcleos grandes y cromatina en grumos y gran cantidad de eosinófilos (Fig. 3). El estudio inmunohistoquímico demostraba una proliferación linfoide de inmunofenotipo T (CD3, CD43 positivo), con positividad para CD30 (Fig. 4), con índice de proliferación (Ki67) del 20%, estableciéndose el diagnóstico histopatológico de infiltración por linfoma T CD30 positivo, compatible con PL tipo A.
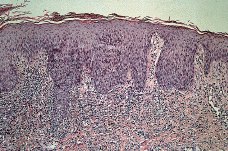
FIG. 2.--Epidermis hiperplásica, con discreta espongiosis y paraqueratosis; ocupación de la dermis reticular y papilar por un denso infiltrado celular con abundantes células linfoides (hematoxilina-eosina).
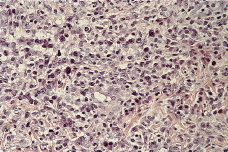
FIG. 3.-- Detalle del infiltrado dérmico con abundantes células atípicas de aspecto linfoide, grandes, con nucléolos evidentes y algunas figuras de mitosis (hematoxilina-eosina).

FIG. 4.-- Las células linfoides grandes muestran positividad para el CD30.
Con este diagnóstico se realizó estudio de extensión para descartar afectación sistémica. El reordenamiento de genes del receptor de células T, realizado por amplificación específica de DNA mediante PCR(s), fue policlonal, tanto en piel como en sangre periférica. El resto de pruebas complementarias realizadas (hemograma, bioquímica, velocidad de sedimentación glomerular, espectro electroforético, radiografía de tórax, sistemático de orina, cuantificación y electroforesis de inmunoglobulinas) fueron normales o presentaron mínimos hallazgos no significativos.
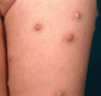
Durante el mes y medio siguiente aparecieron nuevas lesiones en escaso número, que siguieron un curso similar a las previas. A los 2 meses del comienzo la paciente acudió con nuevo brote intenso y generalizado de lesiones asintomáticas, de aproximadamente 1 semana de evolución, distribuidas predominantemente en tronco y raíz de miembros (Fig. 5). No asociaba ninguna sintomatología general. Presentaba un cuadro clínico polimorfo, con lesiones cutáneas papulosas, papulonecróticas y/o nodulares en diferentes estadios evolutivos simultáneamente, algunas con superficie costrosa, otras con signos de regresión y lesiones residuales hiperpigmentadas. El resto de la exploración física fue normal; no presentaba adenopatías ni hepatoesplenomegalia.
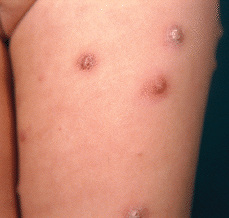
FIG. 5.-- Detalle de lesiones en cara anterior de muslo izquierdo, correspondientes al segundo brote.
Se instauró tratamiento con un corticosteroide tópico (metilprednisolona al 0,1%) y las lesiones remitieron, dejando hiperpigmentación residual; desde entonces, y tras un seguimiento de unos 10 meses desde el inicio del cuadro, la paciente continúa con aparición de nuevas lesiones de forma continua (dos o tres por semana), que evolucionan de forma similar a las anteriores. Actualmente presenta múltiples lesiones papulonodulares en diferente estadio evolutivo, prácticamente asintomáticas, de predominio en extremidades inferiores. Los estudios de control realizados han sido normales hasta la fecha.
DISCUSIÓN
La PL es una enfermedad crónica, caracterizada por un curso clínico benigno, que consiste en una erupción cutánea recurrente y autolimitada de pápulas y pequeños nódulos que frecuentemente se ulceran y curan espontáneamente en 2 a 12 semanas, dejando algunas veces una fina cicatriz atrófica. Las lesiones aparecen predominantemente en tronco y extremidades; su número es muy variable, desde una única lesión hasta cientos de ellas, pudiéndose encontrar en distintos estadios evolutivos simultáneamente. El intervalo de tiempo entre cada brote es variable, sin que se haya identificado un claro factor desencadenante. Su curso clínico es crónico, variando entre 3 meses y 40 años (7-9).
La PL presenta los signos histopatológicos de un linfoma cutáneo de células T. Se distinguen tres variantes histológicas, según el tipo de linfocitos atípicos (7, 10): el tipo A, «histiocítico», presenta una citología similar a la de la enfermedad de Hodgkin, con células grandes, atípicas, CD30 positivas mezcladas con linfocitos pequeños, eosinófilos y neutrófilos; el tipo B, parecido a la micosis fungoide, se caracteriza por un infiltrado en banda de células linfoides atípicas con epidermotropismo, y el tipo C, similar al linfoma anaplásico de células grandes, que está constituido por capas de células T atípicas, grandes CD30 positivas. Estas variantes pueden verse a la vez en un mismo paciente, en lesiones diferentes pero simultáneas. El cuadro histopatológico no tiene significado pronóstico.
A pesar de la histología alarmante, la mayoría de los pacientes con PL presentan un curso clínico benigno, con resolución espontánea; sin embargo, en el 10-20% de los pacientes la PL es precedida, coincidente o seguida de otro tipo de linfoma (11-14), más frecuentemente micosis fungoide (15,16), enfermedad de Hodgkin (17) o linfoma anaplásico de células grandes (18). Un reordenamiento monoclonal de genes del receptor de células T está presente en el 60-70% de los pacientes (10, 19-23).
Aunque existe el acuerdo de que todos los pacientes con PL requieren observación durante largos períodos de tiempo; dado el riesgo de afectación sistémica, existen pocas líneas establecidas de seguimiento. Se considera que el desarrollo de linfoma maligno puede ser reconocido por aumento o persistencia de lesiones cutáneas, por presencia de adenopatías periféricas o de linfocitos atípicos circulantes, por ello se aconseja descartar estos hallazgos al menos una vez al año. Además, ante lesiones cutáneas sospechosas y/o ganglios linfáticos aumentados debe realizarse biopsia para estudio histológico, inmunopatológico y citogenético o reordenamiento genético.
A pesar de la tendencia a la regresión espontánea de las lesiones, se suele instaurar tratamiento para intentar prevenir o limitar la cicatrización, para controlar los síntomas o para disminuir la frecuencia de las recurrencias. Sin embargo, no hay evidencia de que el tratamiento modifique la historia natural de la enfermedad o disminuya el riesgo de degeneración maligna (24).
La mayoría de los casos de PL ocurren en la edad adulta (adultos jóvenes), aunque han sido descritos casos en niños. Thomas y cols. (5) publicaron en 1998 el caso de una niña de 12 años y realizaron una revisión de la literatura inglesa, encontrando sólo 35 pacientes menores de 20 años, apareciendo con mayor frecuencia en la primera década de la vida. El caso de aparición más precoz es el de un niño que comenzó a presentar lesiones a los 8 meses de edad (6).
Debido a su escasa incidencia en la edad infantil, el curso clínico, pronóstico, riesgo de linfoma y recomendaciones de seguimiento no han sido establecidas en niños. La mayoría de los casos de PL en niños, como en el caso de nuestra paciente, son clínica e histológicamente similares a la PL de adultos, pero existen tres patrones especiales (25): a) después de un brote inicial, brotes más leves (tanto en frecuencia como en número de lesiones) hasta que la erupción cesa completamente; b) lesiones localizadas en un área durante años antes de generalizarse, y c) presentación con cientos de lesiones.
La PL en niños se resuelve espontáneamente con mayor probabilidad que en adultos, pero han sido descritos dos casos de comienzo en la niñez, que evolucionaron a otro tipo de linfoma en la edad adul-ta (14, 26).
En cuanto al seguimiento de los niños con PL, Zirbel y cols. (25) sugieren realizar una exploración física exhaustiva cada 6 meses, con atención a crecimiento y desarrollo (herramienta útil para detectar neoplasias ocultas en niños), lesiones cutáneas y adenopatías, y solicitar estudios adicionales, tal como estudio de médula ósea y TAC, sólo si se detectan alteraciones en los exámenes rutinarios. Todos los pacientes con esta enfermedad requieren un seguimiento cuidadoso y continuo a lo largo de toda su vida por la posible aparición de enfermedad sistémica.
Existen muy pocas referencias sobre el tratamiento de la PL en la infancia, ya que, aunque diversas modalidades terapéuticas ensayadas en pacientes adultos son eficaces, su naturaleza invasiva y sus efectos secundarios potenciales limitan su utilización en niños.
Nosotros aportamos un nuevo caso de PL infantil tratado con un corticosteroide tópico que ha presentado dos brotes de lesiones durante un período de seguimiento de unos 10 meses, sin existir ninguna evidencia de afectación sistémica hasta el momento actual.


