





La papulosis linfomatoide (PL) es una dermatosis que se engloba dentro de los procesos linfoproliferativos CD30 positivos de la piel. Se ha descrito su asociación a linfoma de Hogking (LH), así como su progresión a micosis fungoide (MF) y linfoma cutáneo anaplásico de célula grande (LCACG).
ObjetivosInvestigar los hallazgos clínicos, histológicos y la respuesta al tratamiento en un grupo de pacientes con PL.
Material y métodosSe llevó a cabo un estudio retrospectivo, descriptivo y observacional. Se seleccionaron 18 pacientes con diagnóstico histológico confirmado de PL y con un adecuado seguimiento clínico. Se recopilaron los hallazgos histológicos de las biopsias de piel, la forma de presentación, la evolución y la respuesta a los tratamientos utilizados.
ResultadosSe reclutaron un total de 18 pacientes, 10 varones y 8 mujeres. La mayoría de las biopsias, 14 de 18 (78%) mostraban un infiltrado linfocitario en cuña, CD30 positivo, CD3 positivo y CD56 negativo. El tipo histológico más frecuente fue el tipo A, presente en un 83% de las biopsias de los pacientes. La forma clínica de presentación más frecuente fue en forma de pápulas en el tronco (83%). Un 62% de los pacientes sufrió un único brote autorresolutivo. La media de seguimiento fue de 7 años, durante los cuales un 12% de los pacientes desarrolló una micosis fungoide, sin encontrarse otras asociaciones.
DiscusiónExisten pocas series de pacientes con PL publicadas en los últimos años; sin embargo, globalmente los hallazgos descritos en ellas coinciden con las de nuestro grupo de pacientes.
ConclusionesLa PL es un cuadro linfoproliferativo típicamente CD30 positivo que habitualmente tiene un curso benigno con buena respuesta a los tratamientos utilizados.
Lymphomatoid papulosis (LyP) is a CD30+ lymphoproliferative skin disease that has been described in association with Hodgkin lymphoma. It has also been reported to progress to mycosis fungoides or cutaneous anaplastic large-cell lymphoma.
ObjectiveTo study the clinical and histologic features of LyP and response to treatment in a patient series.
Materials and methodsFor this retrospective, descriptive, observational study of patients with histologically confirmed LyP and sufficient follow-up data on record, we extracted histologic findings on skin biopsy, clinical presentation, clinical course, and response to treatments.
ResultsEighteen patients (10 male, 8 female) were identified. Most biopsies (14/18, 78%) showed a wedge-shaped lymphocytic infiltrate with CD30+, CD3+, and CD56− cells. A type A histologic pattern was present in the biopsies of 83% of the patients. The most common presentation (83%) consisted of papules on the trunk; for 62% LyP resolved after a single episode. Twelve percent of the patients developed mycosis fungoides (mean follow-up, 7 years); no other associations were noted.
DiscussionAlthough few series of patients with LyP have been published in recent years, the findings reported generally coincide with our observations.
ConclusionLyP is typically a CD30+ lymphoproliferative disorder that usually runs a benign course and responds well to treatment.
La papulosis linfomatoide (PL) es un síndrome linfoproliferativo que afecta a pacientes de edad media en forma de brotes de pápulas o lesiones pápulo-necróticas recurrentes. Sigue un curso benigno y habitualmente es autorresolutiva en 4-6 semanas. Sin embargo, se ha descrito la capacidad de este cuadro de progresar a otros tipos de linfoma cutáneo de células T, así como su asociación a linfoma de Hodgkin (LH), lo que obliga al seguimiento de los pacientes. Por su curso limitado y benigno frecuentemente no se pauta tratamiento, o simplemente se utilizan corticoides tópicos hasta la resolución de las lesiones. Cuando el curso es más prolongado o las lesiones son más extensas se puede utilizar tratamiento con PUVA, metotrexato a dosis bajas o interferón alfa (IFN-α)1–5.
Tradicionalmente la PL se engloba dentro de los procesos cutáneos primarios linfoproliferativos CD30 positivos y se clasifica en tres tipos histológicos. El tipo A o histiocitoide, caracterizado por la presencia de linfocitos grandes y atípicos acompañados de neutrófilos, histiocitos, eosinófilos y linfocitos pequeños. El tipo B, similar a la micosis fungoide (MF), formado por un infiltrado monomorfo de linfocitos pequeños-medianos de núcleo cerebriforme y la tipo C formado por un infiltrado de linfocitos grandes similar a la tipo A, pero representan más del 50% del infiltrado simulando un linfoma cutáneo anaplásico de célula grande (LCACG). Debido al solapamiento histológico de esta entidad con otros procesos linfoproliferativos cutáneos, así como su posible evolución o asociación a procesos malignos, hace que la discusión de si se trata de una dermatosis benigna, premaligna o maligna esté aún sin resolver, aunque en la actualidad está ampliamente aceptado que la PL es un linfoma cutáneo primario de buen pronóstico. En ausencia de criterios moleculares o de inmunohistoquímica para predecir su curso clínico, la correlación clínico-histológica y el seguimiento de los pacientes es indispensable1,6.
La presencia de reordenamiento clonal del receptor de células T (TCR) se encuentra hasta en el 60% de las lesiones de PL, demostrándose el mismo clon en lesiones separadas7,8; sin embargo, algunos autores encuentran dicha clonalidad en los linfocitos pequeños, lo que no permite diferenciarlos de un proceso reactivo9. Recientemente se describió el marcador MUM-1 como posible diferenciador de PL y LCACG10, pero en estudios posteriores fue desestimado este papel11.
En la literatura revisada existen pocas series publicadas de PL y escasos estudios que contrasten hallazgos clínicos con histológicos e inmunohistoquímicos.
ObjetivosNuestros objetivos fueron investigar los hallazgos histológicos e inmunohistoquímicos (IHQ) de las biopsias de piel en un grupo de pacientes con PL, además de describir las formas de presentación clínica de la enfermedad, su evolución, su asociación a otros procesos, su respuesta a los tratamientos utilizados y la posible correlación de estas variables con los hallazgos histológicos.
Material y métodosSe llevó a cabo un estudio retrospectivo, descriptivo y observacional de pacientes con diagnóstico clínico e histológico de PL y con un adecuado seguimiento. Para seleccionar a dichos pacientes se introdujo como criterio de búsqueda «papulosis linfomatoide» en las bases de datos de anatomía patológica, que engloba desde enero de 2000 a mayo de 2010, y en la base de datos de Dermatología desde enero de 1995 a mayo de 2010. Dichos sistemas proporcionaron 44 y 24 pacientes respectivamente, que finalmente quedaron en 18 pacientes y 26 biopsias en total, tras eliminar a los pacientes que no tenían un diagnóstico correcto, no tenían historia clínica, no tenían las preparaciones histológicas o se había perdido el seguimiento.
Se valoró en este grupo de pacientes las siguientes variables clínicas: edad de inicio, sexo, la presentación clínica (tipo de lesiones y localización), la analítica en el momento del diagnóstico, la evolución de las lesiones, los tratamientos recibidos y la respuesta a los mismos. Se describe también la asociación a otros linfomas, cutáneos o no, así como el tiempo de latencia en aparecer dicha asociación. Con respecto a los hallazgos histológicos se estudió el tipo histológico (A, B, C o formas mixtas), la forma del infiltrado (cuña o banda), la existencia de epidermotropismo, la positividad de los marcadores CD30 (marcador de linfocito T y B activado y de célula de Hodgkin), CD3 (marcador de linfocito T) y CD56 (marcador de células NK) y su intensidad, y la existencia de reordenamiento clonal del TCR en los pacientes en que se realizó. La intensidad de los marcadores de IHQ se graduó en cinco intensidades (+++, ++,+,+-,-) de acuerdo al consenso de dos observadores. Dichos marcadores se realizaron con los siguientes reactivos:
- -
CD3: anticuerpo policlonal de conejo (Dako®, España). IR-503 Flex.
- -
CD30: anticuerpo monoclonal de ratón (Dako®, España). IR-602 Flex.
- -
CD56: anticuerpo monoclonal sintético diluido 1/25. Clon IB6. (Master diagnostics®, España).
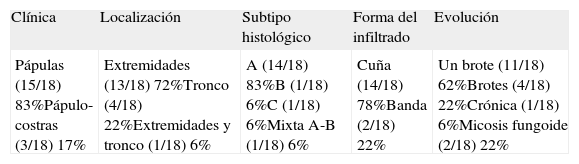
Se resumen en las tablas 1 y 2. Se recopilaron un total de 18 pacientes, de los cuales 10 eran varones y 8 mujeres. La edad media de la muestra fue de 42,7 años, con un rango de 7 a 70 años. Respecto a la forma clínica de comienzo de la enfermedad lo más frecuente fue en forma de pápulas en el 83% de los pacientes (15 de 18), y en forma de pápulo-costras en el resto de los pacientes (3 de 18 [fig. 1]). La localización más habitual de las lesiones fue por orden de frecuencia fue las extremidades en el 72% (13 de 18), el tronco en el 22% (4 de 18) y las extremidades y el tronco en el 6% (1 de 18).
Resumen de los hallazgos clínicos e histológicos de los pacientes
| Clínica | Localización | Subtipo histológico | Forma del infiltrado | Evolución |
| Pápulas (15/18) 83%Pápulo-costras (3/18) 17% | Extremidades (13/18) 72%Tronco (4/18) 22%Extremidades y tronco (1/18) 6% | A (14/18) 83%B (1/18) 6%C (1/18) 6%Mixta A-B (1/18) 6% | Cuña (14/18) 78%Banda (2/18) 22% | Un brote (11/18) 62%Brotes (4/18) 22%Crónica (1/18) 6%Micosis fungoide (2/18) 22% |

Todos los pacientes mostraron al diagnóstico hemograma y bioquímica, incluida LDH, normales, y en los 5 en los que se determinó la beta-2-microglobulina ésta también fue normal. La media de seguimiento de los pacientes después del diagnóstico fue de 7 años, con un rango de 2 a 22 años. En este tiempo la evolución de los casos fue como sigue: la mayoría de los pacientes (11 de 18, un 62%), experimentó un único brote que se resolvió sin tratamiento o con tratamiento tópico. Un 22% (4 de 18) sufrió brotes repetidos a los largo de años, un 12% (2 de 18) desarrolló una MF asociada o comenzó con coexistencia de ambos tipos de linfoma cutáneo de células T y uno de los pacientes permaneció afectado de forma crónica sin respuesta a los tratamientos. Los pacientes que sufrieron brotes repetidos a lo largo de años fueron tratados con PUVA en cada brote con buena respuesta. En el paciente afecto de forma crónica, y en el paciente con coexistencia de lesiones de MF y PL se empleó PUVA en combinación con metotrexato, con control parcial de las lesiones cutáneas.
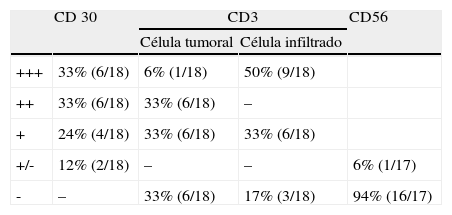
Se reclutaron 26 biopsias en un total de 18 pacientes, ya que algunos tenían biopsias repetidas durante su seguimiento. La mayoría de las lesiones mostraron infiltrado en cuña (78%) frente a banda (22%). El tipo histológico más frecuente encontrado en el 83% de los pacientes fue el tipo A (15 de 18 [fig. 2]), frente al tipo B, C y mixto A y B, encontrados cada uno de ellos en un paciente respectivamente. Todas las muestras tenían positividad para CD30, en la mayoría de los casos moderada o intensa (fig. 3). Respecto a la positividad del CD3 fue más intensa en las células del infiltrado acompañante que en las propias células tumorales (fig. 4), que también se mostraron CD3 positivas, aunque en menor intensidad. Fue llamativo que entre las células acompañantes de las tumorales había positividad para CD3 en un grupo de células y en otro no, pese a su similar morfología, como si se tratara de dos poblaciones celulares diferentes. No se encontró ninguna positividad para CD56, y solo en una de las muestras se clasificó CD56 como dudoso (fig. 5). Solo se encontró epidermotropismo en la forma B y mixta A-B. El reordenamiento del TCR fue negativo en los 9 pacientes en los que se había realizado. Curiosamente en los pacientes en los que había biopsias repetidas (un total de tres pacientes y 7 biopsias), los hallazgos encontrados coincidían y no se detectaron variaciones entre ellas.
.")
.")
.")
.")
La tabla 1 resume las características clínicas de los pacientes y el tipo histológico de PL, sin encontrarse ninguna aparente correlación entre las variables clínicas estudiadas y su clasificación histológica. Debido al pequeño tamaño de la muestra no se pudo aplicar ningún test estadístico con adecuada significación para poder estudiar las variables descritas.
DiscusiónLos hallazgos epidemiólogicos y la clínica de nuestro grupo de pacientes coincide con la literatura publicada2,3; así, la PL se muestra como un proceso linfoproliferativo de ligero predominio en el varón y con una edad media de presentación en torno a los 40-45 años. La mayoría se inician en forma de pápulas o pápulo-costras en los miembros; sin embargo, existen muchas formas descritas de presentación vesiculosas, formas agminadas, formas úlcero-necróticas, etc.1.
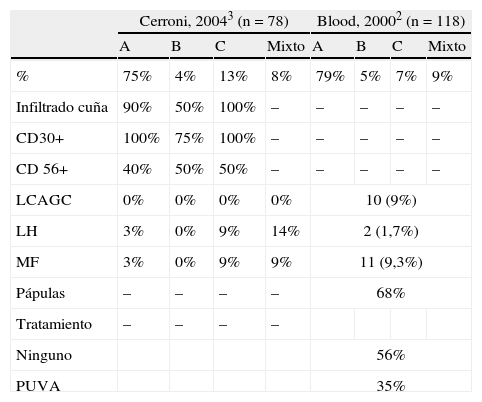
En la literatura revisada de los últimos 10 años solo existen dos series publicadas en las que se estudien los hallazgos clínicos e histológicos, así como sus asociaciones a otras enfermedades en pacientes con PL2,3 (se resumen en la tabla 3). En la literatura española existe además una serie de 9 pacientes pediátricos con PL publicada recientemente4. En la serie de Cerroni publicada en 2004 se describe la histología de las lesiones. En este trabajo se encuentra, al igual que en el nuestro, un claro predominio del tipo A, presente en un 75% de los pacientes. Los tipos B y C se muestran como menos frecuentes, con un 4 y 13% respectivamente, de nuevo similar a nuestra serie de pacientes con un 6% ambas. La mayoría de los infiltrados se describen como en cuña, salvo en los pacientes con el tipo B, que lógicamente tiende a describirse como en banda al ser MF-like. De forma similar, en la serie publicada por Bennek et al. en 20002, de 118 pacientes, la gran mayoría fueron de tipo A, con un 79%, y el resto fueron B, C o formas mixtas con un 5,7 y 9% respectivamente. En este último trabajo no describen los hallazgos de IHQ.
Resumen de los hallazgos de las dos series de pacientes con papulosis linfomatoide publicadas desde el año 2000
| Cerroni, 20043 (n = 78) | Blood, 20002 (n = 118) | |||||||
| A | B | C | Mixto | A | B | C | Mixto | |
| % | 75% | 4% | 13% | 8% | 79% | 5% | 7% | 9% |
| Infiltrado cuña | 90% | 50% | 100% | – | – | – | – | – |
| CD30+ | 100% | 75% | 100% | – | – | – | – | – |
| CD 56+ | 40% | 50% | 50% | – | – | – | – | – |
| LCAGC | 0% | 0% | 0% | 0% | 10 (9%) | |||
| LH | 3% | 0% | 9% | 14% | 2 (1,7%) | |||
| MF | 3% | 0% | 9% | 9% | 11 (9,3%) | |||
| Pápulas | – | – | – | – | 68% | |||
| Tratamiento | – | – | – | – | ||||
| Ninguno | 56% | |||||||
| PUVA | 35% | |||||||
LCAGC: linfoma cutáneo anaplásico de célula grande; LH: linfoma Hodgkin; MF: micosis fungoide.
Con respecto a la IHQ de las lesiones de PL es importante recalcar que no permite diferenciarla de otros procesos CD30 positivos. Esta positividad define claramente esta entidad; sin embargo, existen algunas infrecuentes ocasiones en que no se detecta. Las razones por las que una PL es CD30 negativa no están claras y han sido descritas sobre todo en PL de tipo B12. Una posible explicación es que la clínica que se asumió como compatible con una PL fuera en realidad la posteriormente descrita MF papulosa, y por ello negativa para CD3013.
El CD3 es un marcador pan-T, y por tanto positivo en la PL en casi todos los casos. En nuestra serie de pacientes apreciamos que es más intensamente positivo en las células del infiltrado acompañante a las células grandes, sugiriendo la existencia de dos poblaciones T. Sin embargo, este hallazgo no ha sido encontrado en la literatura revisada, por lo que se necesitan más estudios que lo confirmen.
La razón para explorar el CD56 en nuestra muestra de pacientes fue encontrar que en la serie de Cerroni3 fue positivo en prácticamente el 50% de las biopsias de PL. En nuestro trabajo no fue así, y solo en una de las biopsias fue dudoso CD56 positivo. En la literatura revisada esta positividad en la PL es objeto de controversia y tiende a ser descrita, en general, como algo excepcional en la PL14. Las razones para este hallazgo tan discordante entre dos series de pacientes publicadas en los últimos 6 años no están aclaradas, aunque pueden deberse al uso de diferentes diluciones del reactivo; así Cerroni en 2004 lo utilizó a 1/20, Flann en 2006 en una serie de tres PL CD56 positivas lo diluyó a 1/40 y en nuestro trabajo se utilizó a 1/25.
El muestreo del TCR en la PL es útil para diferenciar lesiones reactivas frente a clonales. Sin embargo, su positividad o negatividad son solo orientativas en el contexto del paciente. Es más, el papel de un reordenamiento clonal en una PL no está aclarado: así, Gellrich et al. en 20039 demuestran la existencia de clonalidad en las células CD30 negativas pequeñas del infiltrado acompañante, mientras que otro autor, Steinhoff en 20027, encuentra la clonalidad en las células grandes CD30 positivas.
La clasificación de la PL en tres subtipos histológicos se muestra, en nuestro trabajo y en la literatura revisada2,3, como algo más tradicional que con transcendencia en cuanto a la clínica, el pronóstico o la respuesta al tratamiento. No se encuentra ninguna relación aparente entre el subtipo histológico y las variables estudiadas.
Con respecto a la evolución de la enfermedad la mayoría, en torno a un 60-70% en los diversos trabajos, son cuadros autolimitados. Sin embargo se ha descrito en la PL el riesgo de progresión a MF en un 10-12% de los casos, asociación a LH hasta en un 9% y asociación a LACG hasta en un 9% (tabla 3). En nuestro grupo de pacientes solo se detectó progresión a MF en un 12% de los casos, y no se encontró ninguna de las otras asociaciones. El diagnóstico de una PL posterior al diagnóstico de una MF debe hacerse con mucha precaución, ya que puede tratarse de una transformación a célula grande, y por tanto supone un cambio radical en cuanto a tratamiento y pronóstico de la MF inicial. La descripción de estas asociaciones en pacientes con PL es la que nos obliga al seguimiento clínico periódico de los pacientes y al estudio semestral con analítica con LDH y placa de tórax5.
La mayoría de las PL son autorresolutivas, papulosas y no precisan tratamiento. En caso de que el brote sea más persistente la respuesta al PUVA suele ser buena, circunstancia que pudimos comprobar en un 22% de nuestros pacientes y en el 35% en la serie de Bennek del 20002. Como alternativa eficaz se utiliza el metotrexato a dosis bajas. Es excepcional encontrar casos resistentes, en los que se ha ensayado bexaroteno15, IFN alfa16, 5-fluorouracilo tópico17 y terapia fotodinámica18 con variable respuesta.
Finalmente, podemos concluir que la PL afecta a pacientes de edad media con brotes de pápulas en las extremidades y en el tronco, en su mayoría autorresolutivos, con buena respuesta a los tratamientos utilizados. La histología típica es un infiltrado en cuña de células grandes CD30+, y en raras ocasiones CD56+. Es preciso el seguimiento de los pacientes al haber sido descrita la asociación con otros procesos linfoproliferativos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


