





INTRODUCCION
La alcaptonuria u ocronosis endógena es una enfermedad hereditaria, autosómica recesiva muy poco frecuente. Su prevalencia se estima entre un caso por cada 250.000 o 1.000.000 de habitantes 1,2. Existe una mayor prevalencia en ciertas poblaciones, como la República Dominicana y en la región Piestany de Eslovaquia, donde se estima en 1:25.000 personas 2. La ocronosis endógena se caracteriza por la incapacidad de metabolizar el ácido homogentísico provocada por una insuficiencia de la enzima ácido homogentísico oxidasa (AHGO). El ácido homogentísico (AHG) se oxida y se acumula en los tejidos formando polímeros y generando depósitos de pigmento, de composición desconocida y de un aspecto similar a la melanina. Al microscopio, este depósito presenta un color ocre-amarillento que se sitúa generalmente en relación con fibras de colágeno del tejido conjuntivo con características degenerativas elastósicas. El término de ocronosis endógena surge como contraposición a otros procesos con depósitos de pigmento ocronótico, generalmente de origen medicamentoso (p. ej., ocronosis exógena por hidroquinona). El término alcaptonuria hace referencia a la clásica adopción de un color marrón oscuro de la orina al oxidarse y en presencia de álcalis 2.
Presentamos un caso típico de esta entidad y posteriormente nos centraremos sobre los aspectos clinicoterapéuticos más importantes de ésta.
DESCRIPCION DEL CASO
Varón de 64 años de edad, fototipo III, camionero, con antecedentes de abuso enólico, cólicos renales, carcinoma papilar transicional no invasivo de vejiga desde 1995, hombro doloroso con rotura de tendón supraespinoso derecho, y artralgias en la rodilla derecha de aproximadamente 2 años de evolución. Fue diagnosticado de alcaptonuria en 2002 por la presencia de pigmento en ambas escleróticas, y posterior detección de ácido homogentísico urinario y estudio radiológico de columna lumbar.
El paciente fue remitido al Servicio de Dermatología en julio de 2003 por lesiones costrosas localizadas en el pabellón auricular izquierdo. Se realizó una biopsia cutánea con diagnóstico anatomopatológico de queratosis actínica bowenoide que se trató con crioterapia. A la inspección cutánea el paciente presentaba un número elevado de pequeñas pigmentaciones negro-azuladas, moteadas, situadas en el borde lateral del segundo dedo de ambas manos, así como algunas de forma aislada en palmas, y en dorso de dedos (fig. 1). También se podía ver una pigmentación moteada difusa, de color azul-grisáceo, en ambos pabellones auriculares, sobre las zonas cartilaginosas, que eran más ostensibles en la zona de las conchas auriculares y antihélix.
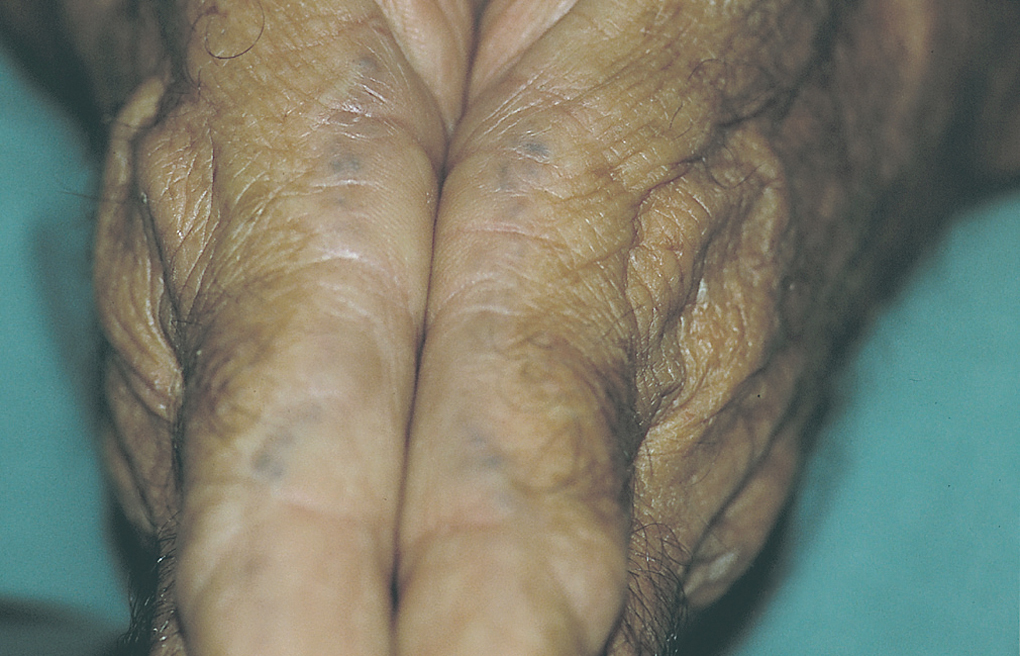
Fig. 1.--Máculas negro azuladas puntiformes en el borde lateral de ambos dedos.
En ambas escleróticas se apreciaban unas pigmentaciones triangulares de color negruzco-azulado-marronáceo situadas entre la córnea y cantos palpebrales. Estas pigmentaciones son características de esta enfermedad y constituyen el signo de Osler (fig. 2). No se apreciaron lesiones en la mucosa oral, sobre zonas tendinosas o uñas. El paciente refería orina de color normal, pero en la infancia había observado que su orina dejaba manchas oscuras en la ropa interior.
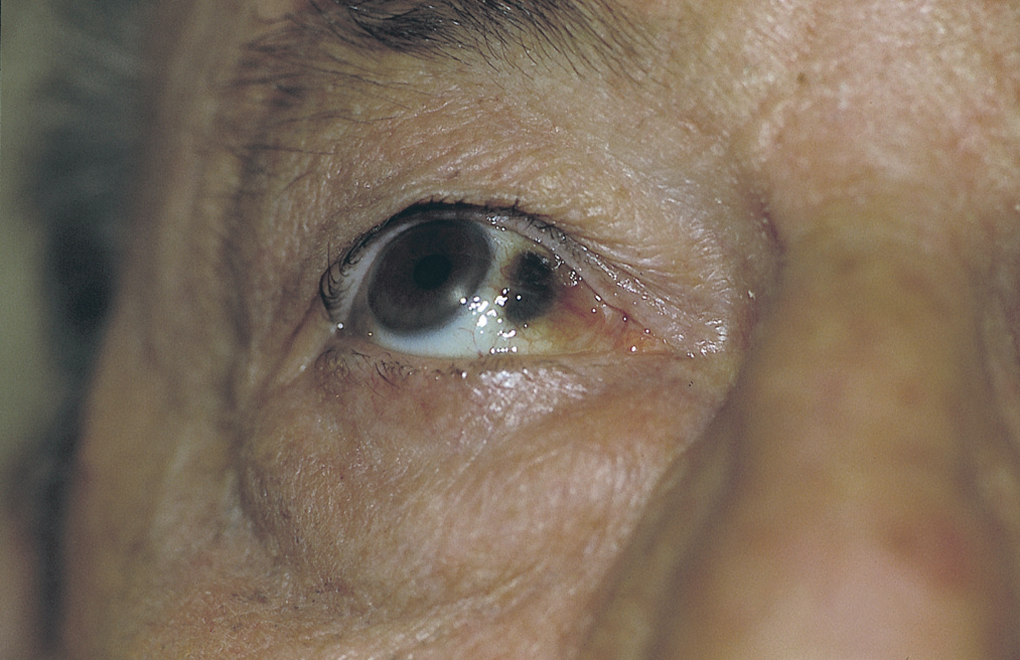
Fig. 2.--Signo de Osler o pigmentación negruzca de la esclerótica.
Se tomó, una biopsia de la cara lateral de uno de los índices afectados, y se observó una epidermis discretamente hiperqueratósica y acantósica. En la dermis superficial había una importante elastosis asociada a depósitos de material acelular de color ocre rodeado de una degeneración basófila del colágeno (figs. 3 y 4).
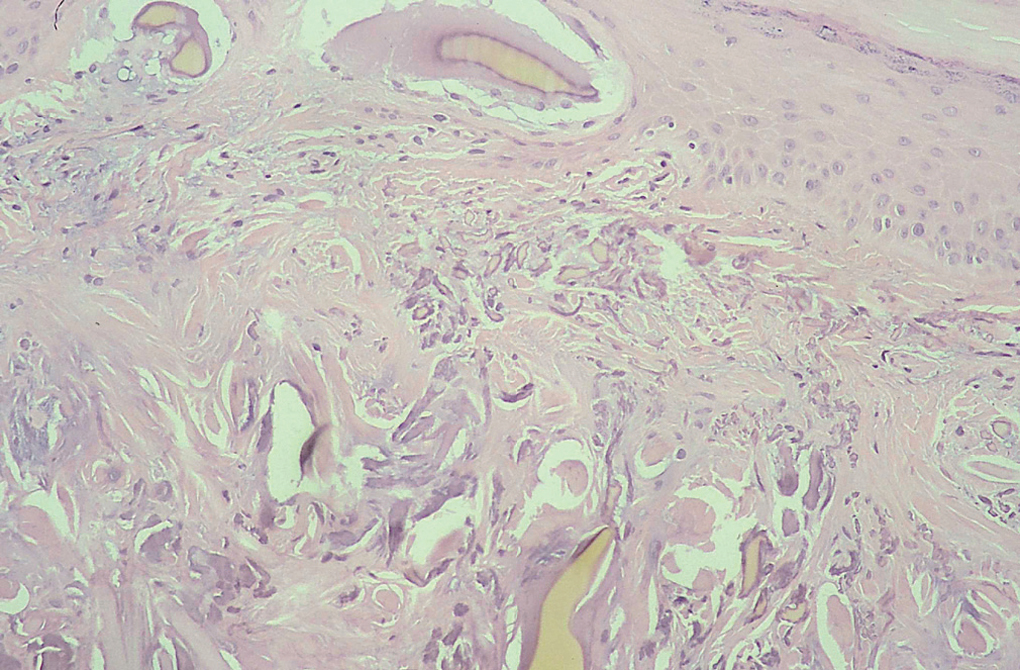
Fig. 3.--Fibras de colágeno irregulares con bordes dentados y fracturadas en relación con cúmulos alargados de pigmento ocre de tamaño grande y pequeño. Algunos en íntima relación con la dermis papilar y epidermis con aspecto de estar en situación previa a su eliminación transepidérmica. (Hematoxilina-eosina, x100.)
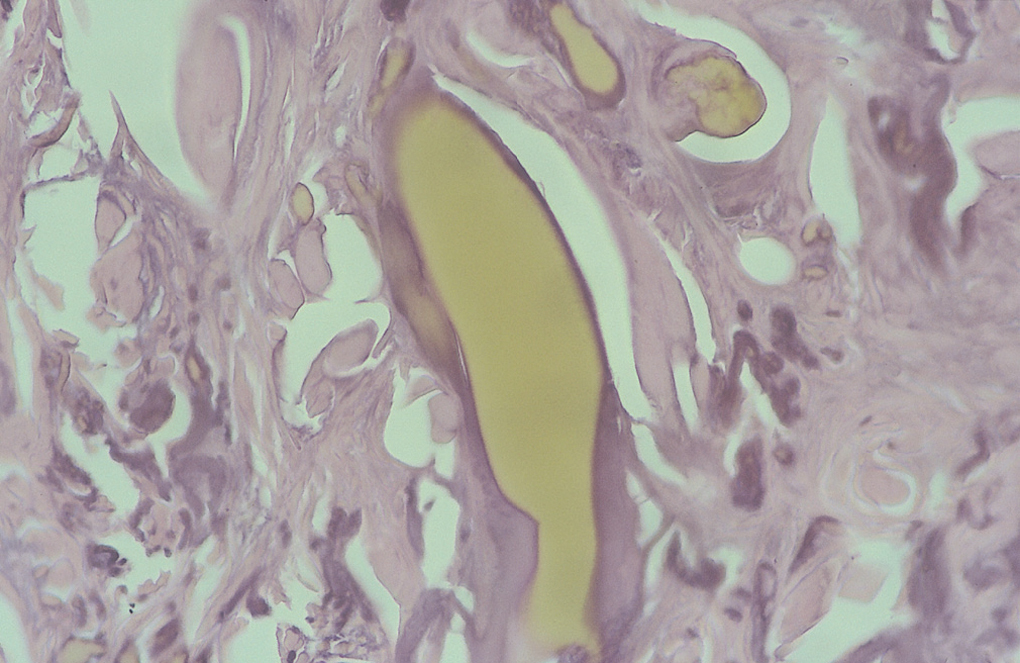
Fig. 4.--Detalle de cúmulos de pigmento de diverso tamaño, el más grande con la forma alargada típica y otras de menor tamaño en íntima relación con las fibras de colágeno de aspecto degenerativo elastótico. (Hematoxilina-eosina, x400.)
La hematimetría, recuento, fórmula, velocidad de sedimentación globular, y perfil bioquímico fueron normales o negativos. El perfil hepático mostró un leve aumento de transaminasas y gammaglutamiltransferasa, y en la ecografía se confirmó una esteatosis hepática de probable origen alcohólico. En orina de 24 h se demostró la presencia de ácido homogentísico muy elevado (5.616,0 mg/24 h [N ≤ 10,0 mg/24 h]). En el estudio radiográfico de la columna lumbar se apreciaron imágenes de osteoporosis difusa, calcificación del ligamento vertebral anterior, osteofitosis anterior, disminución de la altura de las vértebras lumbares y calcificación de todos los discos intervertebrales (fig. 5). La radiografía de la rodilla izquierda mostraba fenómenos de calcificación articular. Se realizó un estudio gammagráfico que demostró la presencia de un refuerzo intervertebral lumbar e imágenes de hipercaptación en la articulación del hombro y de la rodilla. Estos hallazgos gammagráficos en este paciente han sido previamente publicados por Cortés-Hernández et al 3. Se instauró tratamiento con complementos de vitamina C, (1.000 mg/día) y el paciente manifestó una discreta mejoría de sus síntomas articulares.
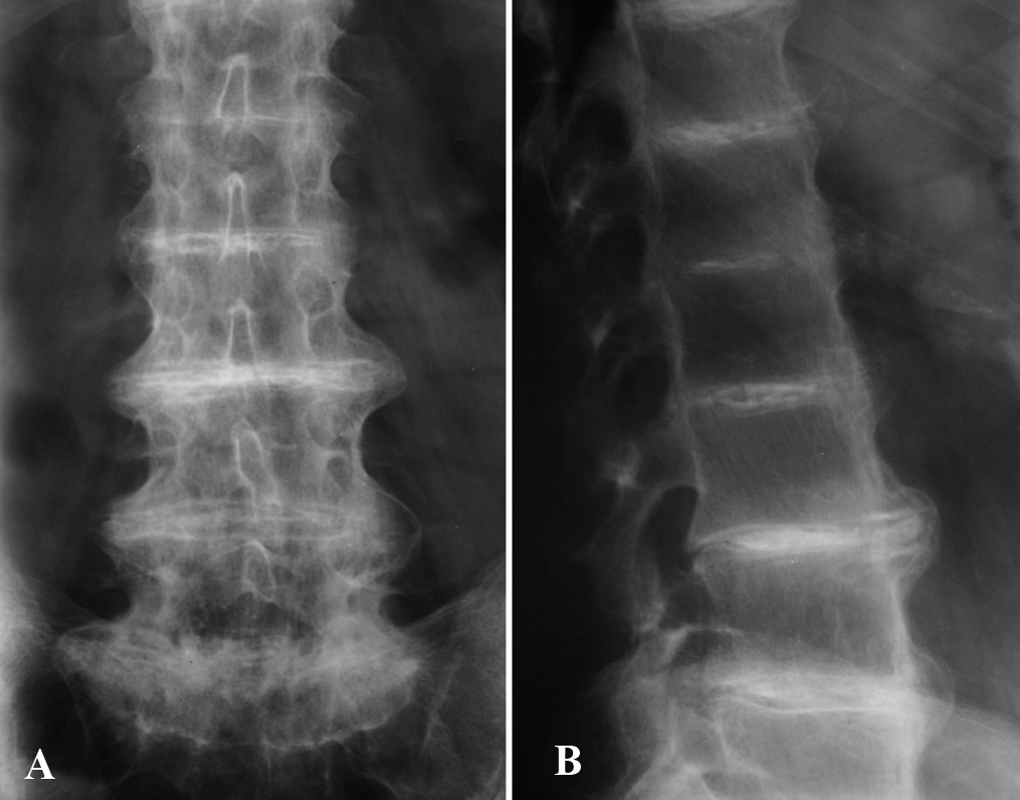
Fig. 5.--Radiografía simple anteroposterior (A) y lateral (B). Estrechamiento y calcificación de las articulaciones intervertebrales. Pérdida de la lordosis fisiológica.
COMENTARIO
La prevalencia de la alcaptonuria es desconocida en España. El gen responsable ha sido localizado en la región 3q21-q23 del cromosoma 3 4. La AHGO se expresa principalmente en el hígado, el riñón y la próstata. El déficit de actividad de la AHGO impide la transformación de AHG en ácido maleilacetoacético. El exceso de AHG, se oxida, se transforma y polimeriza formando el pigmento ocronótico 2,5. El pigmento de la ocronosis tiene características similares a la melanina y tiene afinidad por el colágeno de la piel y el cartílago 6. Se desconoce si este pigmento afecta a la formación de colágeno o si se une al colágeno previamente afectado. En nuestro caso, la deposición de pigmento en una zona atípica para daño actínico apoya más esta primera posibilidad. Sin embargo, este pigmento se describe clínicamente más en zonas de piel fotoexpuesta. También se observa en zonas de piel con mayor densidad de glándulas sudoríparas, lo cual no acontece en nuestro caso 1.
En estudios microscópicos de la piel, el pigmento forma depósitos dérmicos, de color pardo-amarillento, redondeados y alargados en forma de plátano y en estrecha relación con masas de colágeno degenerado. En el caso de la biopsia de nuestro paciente se apreciaron claramente los hallazgos más característicos de esta enfermedad: fibras de colágeno irregulares con bordes dentados y fracturadas en relación con cúmulos redondeados y alargados de pigmento amarillento de diversos tamaños. Sin embargo, no se observaron macrófagos cargados de pigmento en el interior de células endoteliales en células secretoras, ni en la membrana basal de glándulas sudoríparas, hecho descrito también en algunos casos de alcaptonuria. Asimismo se apreciaban grandes depósitos en dermis papilar, lo que indica una probable tendencia a la eliminación transepidérmica en nuestro caso, tal y como se ha descrito en casos de ocronosis exógena 7. Sin embargo, en esta última las fibras ocres son de pequeño tamaño, están dispuestas de forma dispersa en toda la dermis y no se describe la presencia de grandes fibras ocres en la dermis superior. Al igual que en la forma endógena, también se aprecian pequeños gránulos de pigmento ocronótico en el interior de macrófagos. En los casos inducidos por hidroquinona los macrófagos cargados de pigmento están situados en la dermis superior y habitualmente asociados a escasa melanina en la capa basal epidérmica. En los casos inducidos por antipalúdicos estos macrófagos se sitúan a nivel perivascular y perianexial 7. La determinación de AHG en orina se considera patognomónica de esta enfermedad (valores normales en orina: < 0,01 mmol de AHG por mol de creatinina o < 10,0 mg en orina de 24 h) 8.
Aunque los primeros signos de esta enfermedad se pueden observar desde edades tempranas, generalmente pasan desapercibidos. Al nacimiento se aprecian orinas oscuras, que normalmente no lo son a la micción inmediata, pero sí al secarse en el pañal, o cuando se limpian los pañales con jabón. También se observa cerumen oscuro negruzco. Durante la pubertad se puede apreciar en ocasiones una pigmentación axilar. Entre los 20 y los 40 años se suele observar pigmentación azul-grisácea mal delimitada en los pabellones auriculares y pigmentación pardo-grisácea de escleróticas (signo de Osler). Otros lugares habituales de pigmentación son la nariz, la frente, las mejillas, el dorso de las manos, las axilas y los genitales. Al igual que en nuestro caso, también se describen pigmentaciones gris-azuladas palmoplantares que podrían ser más características en pacientes con piel más oscura, donde el resto de pigmentaciones cutáneas son menos apreciables 9, aunque es posible que sea un hallazgo frecuente poco descrito 10. En ocasiones, si la piel es muy fina o presenta claros signos de daño actínico, se puede observar la pigmentación de tendones y cartílagos. También se describe la presencia de pigmento en las uñas y mucosas, incluso en la vaginal 11. La pigmentación cutánea se puede acentuar y acelerar en casos de insuficiencia renal como consecuencia de una disminución de la secreción tubular activa del AHG 1,5,8.
La artropatía asociada a la ocronosis es de tipo degenerativo y carácter progresivo 2. Afecta principalmente al esqueleto axial y grandes articulaciones. Los síntomas musculoesqueléticos comienzan en forma de rigidez y dolor lumbar en un 50 % de los casos antes de los 30 años de edad y en casi todos los pacientes antes de los 40 7. Ocasionalmente se inicia rotura brusca del núcleo pulposo o en forma de monoartritis 8. El 50 % de los pacientes precisan prótesis articulares entre los 55 y 60 años de edad. La afectación tendinosa es frecuente (57 %). El depósito de pigmento en tendones conlleva su engrosamiento, y ocasiona tendinitis, calcificación y rotura de éstos 8. Es muy característico el engrosamiento y la rotura del tendón de Aquiles 11.
Los hallazgos radiológicos, como en nuestro paciente, son característicos si no patognomónicos 2. Hay un estrechamiento progresivo generalizado, calcificación, osificación, e incluso fusión de los espacios intervertebrales 8. A diferencia de las espondilitis, raramente se afectan las articulaciones sacroilíacas. En casos aislados se han realizado estudios gammagráficos con hallazgos equivalentes a cualquier otra artropatía degenerativa, aunque con algún rasgo característico 3.
Las complicaciones urinarias son más frecuentes en varones, donde es típica la formación de cálculos en próstata. También se generan cálculos renales. En la alcaptonuria se deposita pigmento en las válvulas cardiacas, que origina en ocasiones una enfermedad valvular que requiere recambio protésico 8. Hay indicios que señalan que el depósito de pigmento en los vasos sanguíneos puede acelerar la arteriosclerosis, independientemente de la hipercolesterolemia 8. La afectación del tímpano y de la cadena osicular del oído medio puede causar una disminución de la función auditiva.
Generalmente, las manifestaciones clínicas son suficientes para la sospecha del diagnóstico. La confirmación diagnóstica la conseguimos mediante el estudio histológico, estudio radiográfico, presencia de antecedentes familiares y, como prueba definitiva, se realiza la determinación del ácido homogentísico en orina, la cual permite detectar esta enfermedad desde el nacimiento.
En la actualidad, la ocronosis endógena no tiene cura, aunque podría ser un buen modelo para una futura terapia de reposición enzimática 10. La mayor parte de los tratamientos en los pacientes con ocronosis giran en torno al tratamiento sintomático de las secuelas (analgesia, tratamiento quirúrgico y prótesis). Se han ensayado tratamientos con dietas restrictivas en proteínas y aminoácidos para disminuir la producción de AHG, pero con escaso éxito por lo dificultoso de su cumplimiento a largo plazo, sobre todo en la edad adulta 8,13. Se ha propuesto el empleo de vitamina C, aunque hay dudas con respecto a su eficacia a largo plazo y sus efectos biológicos 14. En estudios preliminares in vitro se ha comprobado que el empleo de otros antioxidantes, como la vitamina E y la N-acetil cisteína interfieren con la polimerización del AHG, por lo que podrían demostrar su utilidad en el futuro 15.
Se ha propuesto también el empleo de nitisintona 8. Este fármaco puede reducir la producción de AHG y se emplea en la tirosinemia tipo I asociada a una dieta de restricción proteica. Se ha empleado, durante un breve periodo y a dosis bajas, en 2 adultos con alcaptonuria, en quienes se obtuvieron resultados analíticos alentadores 8. Sin embargo, hay dudas sobre su utilidad y seguridad a largo plazo.


