



La investigación sobre dianas moleculares en el melanoma sobre las que se pueda actuar farmacológicamente está empezando a dar sus primeros frutos. De todos los fármacos ensayados hasta el momento en pacientes con melanoma diseminado, los que han conseguido mejores resultados son los inhibidores de la mutación V600E de BRAF en los melanomas portadores de la misma, los inhibidores de la actividad tirosin-cinasa de c-Kit en melanomas con mutaciones de este gen y los anticuerpos anti-CTLA-4, inhibidores de los mecanismos de inmunotolerancia. Sin embargo, aún quedan muchos problemas por resolver, como la rápida adquisición de resistencias frente a los dos primeros tipos de fármacos o la falta de biomarcadores predictivos de respuesta frente al último de ellos. En este artículo presentamos una revisión sobre los resultados de los tratamientos contra estas y otras dianas en el melanoma diseminado y lo que parece que podemos esperar del futuro.
Research into molecular targets for drug development in melanoma is starting to bear fruit. Of the drugs tested to date in patients with metastatic melanoma, those that have yielded the best results are V600E BRAF inhibitors in melanomas carrying the V600E mutation; c-kit tyrosine kinase activity inhibitors in melanomas carrying c-kit mutations; and anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) antibodies, which block the mechanisms involved in immune tolerance. Many problems have yet to be resolved in these areas, however, such as the rapid development of resistance to BRAF and c-kit inhibitors and the lack of biomarkers to predict treatment response in the case of CTLA-4 blockers. We review the results of targeted therapy with these and other drugs in metastatic melanoma and discuss what the future holds for this field.
El melanoma cutáneo, una vez diseminado, es una enfermedad altamente resistente a los tratamientos antineoplásicos convencionales. Actualmente la dacarbazina (DTIC) sigue siendo el tratamiento estándar para los enfermos con melanoma metastásico con una tasa de respuesta objetiva únicamente del 10-20% y de respuesta completa menor al 5%, de solo 6-8 meses de duración. Por ello, actualmente se están investigando intensamente nuevas estrategias terapéuticas1,2. Hasta hace relativamente poco tiempo, parecía que todos los intentos de actuar sobre el melanoma diseminado con los nuevos tratamientos frente a dianas terapéuticas que tanto han revolucionado otras áreas de la oncología3 acababan en fracaso. Sin embargo, datos aparecidos muy recientemente en la literatura aportan una luz de esperanza a esta situación.
Los tratamientos frente a dianas terapéuticas actúan inhibiendo de forma selectiva a moléculas, generalmente proteínas, de cuya expresión o sobreexpresión depende de forma específica el crecimiento de la neoplasia problema. Así, una de las características principales del tratamiento antineoplásico mediante fármacos contra dianas terapéuticas sería la especificidad, tanto en la relación fármaco-diana como diana-tumor. La especificidad ofrecería como ventaja el evitar la toxicidad de los tratamientos antineoplásicos convencionales que actúan de forma inespecífica tanto sobre células malignas como sobre células normales. Otra ventaja de la especificidad es la de discriminar entre grupos de pacientes aparentemente iguales. Un ejemplo, ya completamente incorporado a la práctica médica habitual, sería la indicación de trastuzumab para el tratamiento del adenocarcinoma de mama únicamente cuando éste sobreexpresa la proteína HER-24. Aunque esta especificidad tan manifiesta no siempre se logra y, como dermatólogos, somos testimonio de muchos de los efectos secundarios de este nuevo grupo de sustancias5, lo cierto es que cada vez se dispone de terapias más selectivas para cada tipo y subtipo de neoplasia, constituyendo lo que se ha denominado tratamiento oncológico «a la carta».
El número de posibles dianas terapéuticas en el melanoma ha ido aumentando a medida que se ha ido conociendo mejor la biología de este tumor y que se han ido sintetizando diversos fármacos contra alguna de las moléculas implicadas en favorecer su crecimiento (tabla 1)6–8.
Posibles fármacos frente a dianas terapéuticas en el melanoma
| Frente a dianas localizadas en las propias células tumorales |
| Frente a moléculas responsables de estimular el crecimiento y/o impedir la muerte celular |
| - Fármacos frente a moléculas antiapoptóticas (oblimersen, ABT-737, ABT-263, YM155) |
| - Inhibidores de proteínas de la cascada de las MAP cinasas: |
| • Inhibidores de la farnesil transferasa (actúan sobre RAS) (tipifarnib) |
| • Inhibidores de RAF (sorafenib, PLX4032, GSK2118436, RAF-265, XL281) |
| • Inhibidores de MEK (PD0325901, AZD6244, GSK1120212, E6201) |
| - Inhibidores de la vía PI3K/AKT: |
| • Análogos de la rapamicina (actúan sobre mTOR) (temsirolimus/CCI-779) |
| • Inhibidores duales de PI3K y mTOR (SF-1126, NVP-BEZ235, NVP-BGT226, XL765) |
| • Inhibidores de PI3K (PX-866, XL147, NVP-BKM120, GDC-0941, CAL-101) |
| • Inhibidores de AKT (MK-2206, GSK690693) |
| - Inhibidores de c-Kit (imatinib, sunitinib, dasatinib, nilotinib) |
| - Inhibidores de acción pleiotrópica: |
| • Inhibidores del proteasoma (bortezomib e inhibidores de segunda generación) |
| • Inhibidores de deacetilasas de histonas (vorinostat y otros) |
| Frente a moléculas responsables de facilitar la invasión y/o las metástasis |
| - Terapia anti-moléculas de adhesión: |
| • Anticuerpos anti-integrina αvβ3 (etaracizumab) |
| • Inhibidores de la caderina N (ADH-1) |
| • Anticuerpos anti- MCAM/MUC18 (ABX-MA1) |
| Frente a dianas localizadas en estructuras ajenas a las células neoplásicas |
| Fármacos antiangiogénicos |
| - Anticuerpos anti-VEGF (bevacizumab) |
| - Inhibidores de VEGFRs (sunitinib, sorafenib, semaxinib, axatinib) |
| - Antagonistas de las integrinas (cilengitide) |
| - Talidomida y derivados (lenalinomida) |
| Fármacos inhibidores de los mecanismos de inmunotolerancia |
| - Anticuerpos anti CTLA-4 (ipilimumab y tremelimumab) |
Muchas de las moléculas pertenecientes a este grupo son factores de crecimiento, receptores de los mismos o proteínas involucradas en las vías de señalización intracelular, con efecto proproliferativo o antiapoptótico, la mayoría producto de oncogenes6. Las células de melanoma tienen fuertemente activadas estas vías (fig. 1), ya sea a través de mutaciones de genes que codifican proteínas implicadas en las mismas, como a variaciones en los niveles de expresión proteica. Por ello, poseen gran capacidad proliferativa y una resistencia natural a los mecanismos extrínsecos y/o intrínsecos que inducen la muerte celular programada o apoptosis. Algunos de los mecanismos responsables de la alteración de dichas redes de señalización celular son los siguientes: 1) la activación constitutiva de receptores de factores de crecimiento (c-Kit, PDGFR-α, EGFR); 2) la activación de la vía de señalización de las MAP-cinasas (RAS/RAF/MEK/ERK); 3) la activación constitutiva de la vía AKT (PI3K/AKT), favorecida entre otros mecanismos por mutaciones, deleciones o silenciamientos del gen supresor tumoral PTEN; 4) las alteraciones de la red de control del ciclo celular (deleción, silenciamiento o mutación de CDKN2A, amplificación de CDK4 o de CCND1); 5) el deterioro de la actividad transcripcional de la proteína propapoptótica p53, y 6) la sobreexpresión de proteínas antiapoptóticas de la familia de Bcl-2, resultado de las aberraciones en varias de las vías de señalización intracelular mencionadas9–11. Cualquiera de las proteínas situadas en puntos estratégicos de estas vías podría resultar a priori una buena diana molecular para el tratamiento del melanoma diseminado.
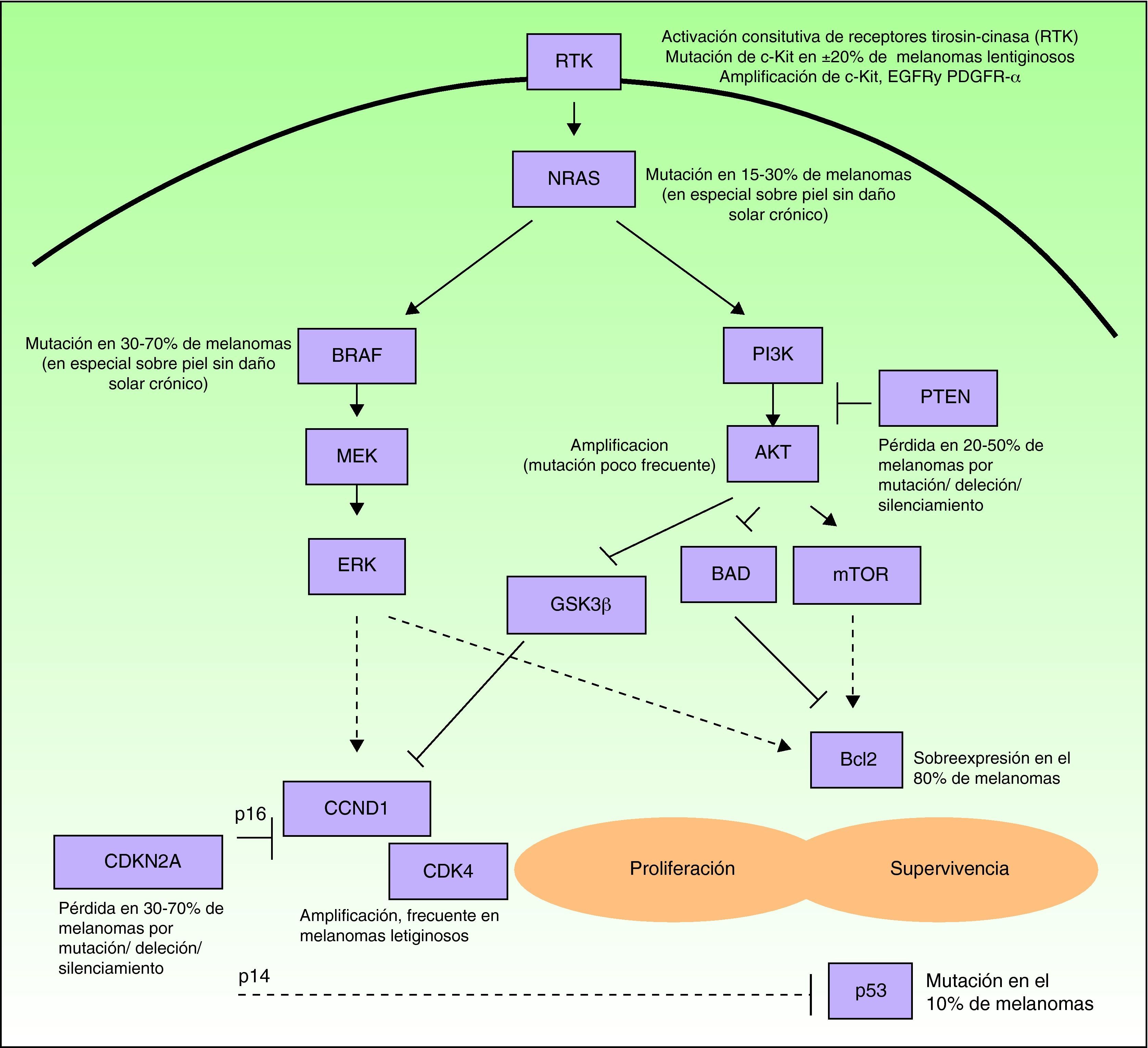
Esquema simplificado de algunas de las principales vías intracelulares implicadas en favorecer la proliferación (estimulación del ciclo celular) y la supervivencia (estimulación de los mecanismos antiapoptóticos y supresión de los mecanismos propapoptóticos) de las células de melanoma. Muchos melanomas poseen una activación constitutiva de receptores de factores de crecimiento con actividad tirosin-cinasa (RTK) (pe, c-Kit, PDGFR-α y/o EGFR), ya sea por amplificaciones o mutaciones de los genes que los codifican. Asímismo, presentan activación de la vía de señalización de las MAP-cinasas (proteín cinasas activadas por mitógenos) (RAS/RAF/MEK/ERK), debida fundamentalmente a mutaciones de NRAS o de BRAF. Con frecuencia, poseen además activada la vía de las fosfatidil-inositol 3 cinasas (PI3K) (PI3K/AKT), de forma secundaria a las mutaciones de NRAS, por amplificación de AKT y, especialmente favorecida por la pérdida del papel inhibidor de PTEN, resultado de mutaciones, deleciones o silenciamientos del gen supresor tumoral PTEN. Por otro lado, en la activación del ciclo celular, contribuyen las deleciones, silenciamientos y mutaciones de CDKN2A, que repercuten en defectos de las 2 proteinas codificadas por este gen (p16 y p14), así como las amplificaciones de CDK4 y CCND1 (ciclina D1). Todo ello influye en el deterioro de la actividad de la proteína propapoptótica p53 (que también puede estar mutada en algunos casos) y la sobreexpresión de proteínas antiapoptóticas de la familia de Bcl-2.
Un concepto importante que ha cambiado la estrategia con la que abordar el tratamiento del melanoma metastásico ha sido el comprobar que el melanoma es un tumor genéticamente heterogéneo, de modo que las alteraciones anteriormente mencionadas varían de unos subgrupos de melanoma a otros. Aunque datos anteriores ya apuntaban en esta dirección, los primeros trabajos extraordinariamente relevantes en este sentido fueron los publicados en 2005 y 2006 por Curtin et al.12,13. Dichos autores dividieron a los melanomas cutáneos y mucosos en los siguientes 4 grupos relacionados con distintos patrones de exposición solar y diferente localización anatómica: 1) melanomas sobre piel sin daño solar crónico (o melanoma relacionado con la exposición solar intermitente); 2) melanomas sobre piel con daño solar crónico (que corresponderían fundamentalmente al melanoma sobre lentigo maligno); 3) melanomas acrales, y 4) melanomas de mucosas. Hallaron que, mientras el 81% de los melanomas en piel sin daño solar crónico tenía mutaciones de BRAF o de NRAS (que eran mutuamente excluyentes), la mayoría de los melanomas de los otros tres grupos, los tres con un patrón histológico lentiginoso, no presentaban mutaciones de estos genes, pero sí amplificaciones de los genes CDK4 y CCND1 (ciclina D1) y/o aberraciones genéticas de c-Kit que incluían mutaciones y amplificaciones.
Estos datos se han ido ampliando con posterioridad, tanto respecto a los tipos de mutaciones o alteraciones que con mayor frecuencia se detectan en los genes mencionados, como en la medida en que pueden combinarse entre sí o con otros trastornos moleculares (p. ej. pérdida de PTEN, amplificación de AKT, etc.) conformando distintos subgrupos de melanoma. De este modo, la terapia mediante fármacos frente a dianas selectivas en el melanoma debería tener en cuenta esta variabilidad y realizarse de forma personalizada, según el perfil genético y de expresión de cada subgrupo de tumores9,11,14,15.
Terapia antisentido frente a Bcl-2. Otras moléculas implicadas en la apoptosis como posibles dianas terapéuticas en el melanomaEsta es una de las primeras estrategias frente a dianas terapéuticas que se han ensayado para el tratamiento del melanoma metastásico. Bcl-2 es una proteína antiapoptótica que está sobreexpresada en muchas neoplasias, concretamente en el 80% de los melanomas, favoreciendo la supervivencia celular. Como terapia antisentido contra Bcl-2 se emplea un fármaco, denominado oblimersen. El primer ensayo clínico fase I-II en el que se combinó oblimersen y DTIC obtuvo unos resultados aceptables que animaron a diseñar un segundo ensayo fase III randomizado con un grupo de pacientes que solo recibía DTIC y un grupo que recibía DTIC más oblimersen. Se incluyeron 771 pacientes. En comparación al grupo que únicamente recibía DTIC, en el grupo que recibía DTIC más oblimersen se observó una mayor tasa de respuestas objetivas (13,5 vs 7,5%; p=0,007) y un aumento del tiempo libre de enfermedad (mediana: 2,6 meses vs 1,6 meses; p<0,001), pero solo una tendencia a un aumento de la supervivencia global (mediana: 9,0 meses vs 7,8 meses; p=0,077). Aunque un análisis retrospectivo indicó que en el subgrupo de pacientes sin elevación de la LDH sérica se obtuvo un beneficio respecto a la supervivencia global (mediana: 11,4 vs 9,7 meses; p=0,02)16, este dato no ha podido ser comprobado en ensayos posteriores. Se ha intentado combinar oblimersen con otros citostáticos, como paclitaxel y temozolamida, sin conseguir mejorar estos resultados, por lo que de momento se ha abandonado esta vía como posible tratamiento del melanoma8,17. Actualmente se están ensayando tratamientos contra otras proteínas antiapoptóticas como ABT-737 y ABT-263 (miméticos de BH3) e YM155 (inhibidor de survivina)17.
Inhibidores de BRAFRAF es una familia de proteínas (ARAF, BRAF y CRAF) que intervienen en la vía de señalización intracelular de las MAP cinasas regulando la proliferación, la diferenciación y la supervivencia celular. BRAF está mutado en el 30-70% de los melanomas, especialmente, como ya se ha comentado, en aquellos sobre piel sin daño solar crónico12,13, que constituyen el grupo más frecuente de melanoma. En más del 90% de los casos la mutación del gen BRAF es siempre la misma, una mutación puntual del exón 15 (cambio de timina por adenina), que causa la sustitución de ácido glutámico por valina en la posición 600 de la proteína BRAF (mutación V600E). Por otro lado, el 15-30% de los melanomas presenta mutaciones de NRAS, que al codificar una proteína situada al inicio de vía de las MAP cinasas (fig. 1), actúan también como activadoras de BRAF. Curiosamente, se han detectado mutaciones de BRAF en el 20% y de NRAS el 80% de los nevus melanocíticos comunes adquiridos y congénitos, de significado biológico aún incierto10. Todo ello ha llevado a pensar que BRAF es una proteína muy importante para la proliferación de las células de origen melanocítico18–20.
Se ha intentado relacionar la presencia de mutaciones de BRAF y NRAS con diversas características clínicas e histopatológicas de los melanomas portadores de las mismas21–23. Aunque aún se precisan más trabajos en este sentido, a modo de resumen, los melanomas con mutaciones de BRAF suelen ser lesiones más pigmentadas, localizadas en el tronco o las extremidades de adultos de edad media (<50 años), con historia de exposición solar en la infancia, escasas efélides y sin queratosis actínicas. Desde el punto de vista histológico, asientan sobre piel con poca elastosis actínica, se asocian con mayor frecuencia a la variante de melanoma de extensión superficial, las células neoplásicas tienden a invadir los estratos altos de la epidermis formando nidos intraepidérmicos (patrón de crecimiento pagetoide), la epidermis afecta suele estar engrosada y bien delimitada respecto a la piel circundante, las células suelen ser redondeadas, grandes y claramente pigmentadas, el Breslow suele ser menor y la tasa de mitosis más baja. En su comportamiento clínico, tienden a metastizar más en los ganglios linfáticos regionales y poseer mejor supervivencia.
SorafenibEl primer fármaco empleado clínicamente para inhibir BRAF ha sido BAY43-9006, llamado también sorafenib. Sorafenib fue diseñado para inhibir la actividad tirosin-cinasa de CRAF, pero pronto se vio que también inhibía a BRAF, tanto a la proteína wild-type (normal o «salvaje») como a la proteína mutada (mutación V600E y otras). Posteriormente, se ha visto que sorafenib es en realidad un inhibidor multicinasa, capaz de inhibir a otras muchas moléculas como VEGFR2 y -3, PDGFR-β, p38 MAPK, FLT3, c-Kit y RET24. En principio, esta cualidad de sorafenib no tendría porque ser un hecho negativo, ya que las células de melanoma poseen varios receptores tirosin-cinasa activados e, incluso, el efecto sobre VEGFR2 y -3 podría tener una acción antiangiogénica. Sin embargo, a pesar de que los experimentos preclínicos, tanto in vitro como en modelos animales, parecían ser alentadores, los resultados de los ensayos clínicos, que se comentan a continuación, no han confirmado la eficacia de sorafenib para el tratamiento del melanoma diseminado18–20.
En el primer ensayo clínico fase II, que incluía 39 pacientes, utilizando únicamente sorafenib, se consiguieron una respuesta completa y 7 respuestas menores. En un segundo ensayo fase I/II, sobre 35 pacientes, al añadir carboplatino y paclitaxel, el número de respuestas parciales aumentó a 11, además de obtenerse 19 respuestas menores. Posteriormente se realizó un ensayo fase II con dos brazos (sorafenib + dacarbazina vs placebo + dacarbacina) en el que se observó un aumento del intervalo libre de enfermedad en el grupo que recibía sorafenib, pero sin mejora en la supervivencia global. Posteriormente se han diseñado otros ensayos fase III, combinando sorafenib con otros citostáticos (temozolamida, carboplatino, placlitaxel) sin conseguir mejorar los resultados. En los ensayos clínicos mencionados, la respuesta no se pudo correlacionar con la presencia de la mutación V600E de BRAF. Se piensa que quizá la diana clínica de sorafenib sea en realidad más VEGFR2 o PDGFR-β que BRAF y, de hecho, actualmente unas de sus principales indicaciones son el tratamiento del carcinoma de células renales y el del carcinoma hepatocelular inoperable donde la angiogénesis tiene un papel más relevante18–20. Algunos estudios preclínicos sugieren que sorafenib sería más efectivo en un grupo pequeño de melanomas portadores de mutaciones de BRAF distintas a la V600E25.
Inhibidores selectivos de BRAF: PLX4032 y otras moléculasDespués del fracaso de sorafenib en el melanoma, se han sintetizado inhibidores más específicos de BRAF y, en concreto de la proteína con la mutación V600E. El primero de los inhibidores específicos de BRAF-V600E que ha llegado a ensayos clínicos es PLX4032, un fármaco de bajo peso molecular, de administración oral.
En el primer ensayo clínico recientemente publicado26 se incluyeron 87 pacientes. Este ensayo se desarrolló en dos fases. En una primera fase I, se reclutaron 55 pacientes, 49 pacientes con melanoma metastásico (con y sin mutaciones de BRAF) y 6 pacientes con otras neoplasias que suelen presentar mutaciones de BRAF (tres de ellos con carcinoma papilar de tiroides con la mutación BRAF/V600E). En una extensión fase II del ensayo, se reclutaron 32 pacientes, todos con melanomas portadores de la mutación V600E de BRAF que recibieron la dosis de PLX4032 establecida como idónea en la primera fase.
Entre los 55 pacientes de la primera fase que recibieron una dosis de PLX4032≥240mg/12h, se encontraban 16 enfermos con melanoma metastásico con la mutación V600E de BRAF. En estos pacientes la tasa de respuesta global fue del 69%, incluyendo una respuesta completa. No se observaron respuestas en ninguno de los pacientes con melanoma metastásico sin mutación V600E de BRAF que, sin embargo, habían recibido una dosis de PLX4032≥240mg/12h. En los tres pacientes con carcinoma papilar de tiroides se obtuvieron respuestas objetivas; uno de ellos mantuvo un intervalo libre de enfermedad de 12 meses y los otros dos, enfermedad estable durante 11 y 13 meses.
La totalidad de los 32 pacientes incluidos en la fase de extensión presentaban melanoma metastásico con mutación BRAF/V600E y recibieron una dosis de PLX4032 de 960mg/12h. Se observó respuesta objetiva en el 81% de los pacientes (26/32), con dos respuestas completas y 24 respuestas parciales. Tal como ya se había observado en la primera fase, se produjeron respuestas en pacientes con metástasis viscerales en localizaciones habitualmente resistentes al tratamiento (como hepáticas, intestinales y óseas), con concentración sérica de LDH elevada o que no habían respondido a otras terapias. La mediana de duración del período libre de enfermedad fue de 7 meses. De los efectos secundarios, el más llamativo, especialmente desde el punto de vista dermatológico, fue la aparición de carcinomas escamosos cutáneos bien diferenciados/queratoacantomas en el 32% de los pacientes.
En conjunto, nunca se han publicado resultados tan buenos en el tratamiento del melanoma metastásico. Sin embargo, dado que el tiempo de seguimiento no es suficiente, aún no se conoce el impacto que estas respuestas tan llamativas puedan tener en la supervivencia global. De hecho, a pesar de haber conseguido una buena respuesta, las recaídas aparecen precozmente, por lo general en un período de 8-12 meses después del tratamiento27. Estos datos sugieren que la resistencia a PLX4032 se desarrolla con facilidad28. Los primeros estudios sobre los mecanismos de resistencia a PLX4032 apuntan a que ésta se debe a la reactivación de la vía de las MAP cinasas, no por aparición de nuevas mutaciones de BRAF, sino por el desarrollo de mutaciones de NRAS, activación de PDGFRβ o sobreexpresión de COT/TPL227,29,30. Para evitar la aparición de resistencias se ha propuesto utilizar PLX4032 en combinación con fármacos inhibidores de otras dianas, como otra molécula de la vía de las MAP cinasas, por ejemplo MEK, o la mencionada COT (ver apartado: Tratamientos combinados).
La posibilidad de inhibir de forma específica la mutación V600E de BRAF ha conducido además a ampliar nuestro conocimiento sobre las interacciones entre BRAF, CRAF y la activación de las vía de las MAP cinasas. Se ha comprobado que en los melanomas que no poseen la mutación V600E de BRAF (por ejemplo, melanomas con mutación de NRAS, pero con BRAF normal o wild-type), la inhibición específica de BRAF induce la formación de dímeros BRAF/CRAF que, paradójicamente, acaban teniendo un efecto activador sobre la vía de las MAP cinasas, y, por tanto, sobre el crecimiento celular. Por ello, es muy importante seleccionar para el tratamiento con estos inhibidores específicos solo a aquellos pacientes con melanomas portadores de la mutación V600E31–35. Por otro lado, este mecanismo podría explicar uno de los efectos secundarios del tratamiento con PLX4032, como es la aparición de carcinomas escamosos cutáneos/queratoacantomas al inducir la proliferación de células epiteliales cutáneas normales que, lógicamente, no poseen de mutaciones de BRAF36.
Otros inhibidores específicos de BRAF, algunos de los cuales están ya siendo evaluados en ensayos clínicos, son GSK2118436 (que inhibe selectivamente la actividad tirosin-cinasa de las proteínas RAF, con mayor potencia respecto a BRAF que a CRAF), RAF-265 (que inhibe intensamente todas las isoformas de RAF, ARAF, BRAF y CRAF, incluyendo la mutación V600E de BRAF, además de VEGFR-2, c-Kit, y PDGFRβ) y XL281 (que actúa también sobre las diferentes RAF cinasas)28.
Inhibidores de c-Kit. Imatinib (STI-571) y otros inhibidores de la actividad tirosin-cinasa de c-KitLa posibilidad de que c-Kit fuera una diana terapéutica en el melanoma ya hace tiempo que se baraja. De hecho, c-Kit es una proteína que actúa como un receptor de factor de crecimiento fundamental de los melanocitos epidérmicos y posee un papel primordial en la diferenciación y migración de las células melanocíticas durante el desarrollo embrionario37. En concordancia, muchos melanomas expresan c-Kit. Sin embargo, algunos datos clínicos y experimentales apuntaban en contra de esta hipótesis hasta hace pocos años. Por un lado, se había demostrado que la expresión de c-Kit disminuía con la progresión tumoral de muchos melanomas38,39 y que, en algunas líneas celulares, la pérdida de este factor de crecimiento se relacionaba paradójicamente con un aumento de la capacidad metastatizante40,41. Por otro lado, los primeros ensayos clínicos realizados en pacientes con melanoma metastásico, empleando fármacos inhibidores de c-Kit, como imatinib (STI571), obtuvieron resultados muy desalentadores42,43.
A pesar de que existían evidencias experimentales y clínicas previas de respuestas aisladas a imatinib y de mutaciones muy ocasionales en el exón 11 de c-Kit en algunas células de melanoma44–48, no fue hasta el trabajo publicado por Curtin et al. en 200613 cuando se estableció que un 39, 36 y 28% de determinados tipos de melanoma, de por sí poco frecuentes (melanoma de mucosas, melanoma acral y melanoma sobre piel con daño actínico crónico, respectivamente), se encontraban aberraciones genéticas de c-Kit que convertían a la proteína codificada por este gen en una posible diana para su tratamiento13. Dichas alteraciones no se han hallado en el melanoma uveal. La baja representación de estos tipos de melanoma en las series de melanoma metastásico, explicarían la falta de respuesta observada en los ensayos clínicos anteriormente referidos49. En un ensayo clínico, diseñado antes de conocer estos datos, pero publicado en el 2008, el único paciente que había respondido a imatinib era portador de un melanoma lentiginoso acral50.
Las alteraciones genéticas de c-Kit incluyen mutaciones y amplificaciones de este gen. Este dato es importante porque no necesariamente los dos tipos de alteraciones tienen porqué tener el mismo significado biológico. Del 39-28% global de aberraciones genéticas de c-Kit, solo un 15-38%, un 8-23% y un 0-17% (de los melanomas mucosos, acrales y sobre piel con daño actínico crónico, respectivamente) corresponden a mutaciones. La mayoría de las mutaciones, tal como ocurre en los GIST (Gastrointestinal Stromal Tumors), neoplasias en las que son típicas las mutaciones de c-Kit, están localizadas en el exón 11. Sin embargo, existe una proporción mayor que en los GIST de mutaciones localizadas en los exones 13, 17 y 18, que precisamente corresponden a mutaciones asociadas a resistencia a imatinib. Por otro lado, en el melanoma se ha descrito la coexistencia de mutaciones y amplificaciones de c-Kit, algo no habitual en los GIST. Los estudios realizados hasta el momento parecen indicar que no siempre existe una relación entre la positividad o negatividad inmunohistoquímica para c-Kit y la presencia o ausencia de aberraciones genéticas. Por tanto, los estudios moleculares parecen imprescindibles para establecer la existencia de alteraciones de este gen14,51–54.
Además de imatinib, existen ya en el mercado distintos fármacos capaces de inhibir la actividad tirosin-cinasa de c-Kit como sunitinib, dasatinib y nilotinib3. La capacidad de estos fármacos para inducir la regresión de melanomas portadores de mutaciones activadoras de c-Kit se ha demostrado en varios casos clínicos publicados de forma aislada2,55–58 y en series cortas51. Actualmente se están llevando a cabo varios ensayos fase II con distintos fármacos capaces de inhibir c-Kit en pacientes afectos de melanoma diseminado portador de aberraciones del gen. Estos ensayos permitirán evaluar entre otros los aspectos siguientes: 1) si la distinta sensibilidad o resistencia de las diferentes mutaciones de c-Kit frente a los distintos fármacos inhibidores de c-Kit tiene una traducción clínica relevante, de forma que se tenga que adaptar el fármaco a emplear al tipo de mutación que se halle en cada caso; 2) qué inhibidores actúan de forma eficaz sobre las metástasis del sistema nervioso central; 3) si existen variaciones en la respuesta clínica dependiendo de que el melanoma posea una mutación activadora de c-Kit, únicamente amplificaciones, o la combinación de mutación activadora junto a amplificación (a este respecto, en un estudio realizado con imatinib, se obtuvo una tasa de respuesta del 50% en los pacientes con melanomas con mutaciones de c-Kit, pero ninguna en los portadores únicamente de amplificación del gen59) y 4) cuáles son los mecanismos de desarrollo de resistencias a los inhibidores de c-Kit. Sabemos que en los GIST esto ocurre habitualmente por la aparición de mutaciones adicionales de c-Kit, pero aún no se conoce que es lo que se puede esperar del melanoma8,52,53.
Inhibidores de RASLas isoformas del oncogén RAS comprenden KRAS, HRAS y NRAS. Un 15-30% de los melanomas poseen mutaciones de NRAS. Las mutaciones activadores de RAS estimulan la vía de las MAP cinasas, pero también la vía de PI3K/AKT (fig. 1) y otras. Los primeros fármacos utilizados para intentar inhibir la vía de las MAP cinasas fueron los inhibidores de la farnesil transferasa de RAS (tipifarnib o R115777). Se realizó un solo ensayo fase II de un único brazo que se cerró por falta de respuesta. Sin embargo, ninguno de los pacientes fue seleccionado en función de la presencia de mutaciones de NRAS. Hay alguna evidencia de que los antagonistas de RAS podrían aumentar el efecto de la quimioterapia, pero este enfoque no se ha intentado clínicamente en el melanoma. Se debería trabajar más en la síntesis de nuevos fármacos capaces de inhibir RAS de forma más efectiva8. Otra alternativa sería inhibir de forma combinada las dos vías principales que se activan como consecuencia de la activación de RAS, como son la vía de las MAP cinasas y la vía PI3K/AkT14,60 (ver apartado: Tratamientos combinados).
Inhibidores de MEKMEK es un proteína de la vía de las MAP cinasas, situada de forma descendente después de BRAF. Se han sintetizado diversos inhibidores de MEK (PD0325901, AZD6244, GSK1120212, E6201). Con los resultados obtenidos en los primeros ensayos fase I o fase II, no parece que estos agentes farmacológicos puedan ser efectivos como fármacos únicos en el tratamiento del melanoma. Sin embargo, existen muchos estudios preclínicos que sugieren que serían una buena alternativa para los tratamientos combinados, tanto para evitar resistencias en el uso de fármacos dirigidos contra la mutación BRAF/V600E, como para el tratamiento de mutaciones de BRAF distintas a la V600E o mutaciones de NRAS, especialmente si se asocian a inhibidores de la vía de la PI3K/AKT8,14,54,61 (ver apartado: Tratamientos combinados).
Inhibidores de la vía PI3K/AKTLa vía PI3K/AKT también se estimula de forma descendente a partir de la activación de RAS. En el melanoma suele estar intensamente activada, tanto por mutaciones del oncogen NRAS (15-30% de los casos), como por mutaciones (10-20% de los melanomas) o silenciamientos del gen supresor tumoral PTEN, que impiden el efecto fisiológico regulador de PTEN sobre la vía, o amplificaciones de AKT. Recientemente se ha identificado una mutación puntual poco frecuente de AKT3 E17K, activadora de AKT (fig. 1)8,10,14,54,61. Como agentes inhibidores de la vía PI3K/AKT se han utilizado diferentes derivados de la rapamicina (CCI-779 o temsirolimus). Estas moléculas actúan sobre mTOR, molécula situada en la vía de forma descendente después de AKT/PKB (fig. 1). Existen también inhibidores duales de PI3K y mTOR, de PI3K y de AKT62. Aunque los resultados clínicos de estos fármacos en ensayos fase II no han sido buenos, existen diversos autores que proponen su utilidad en tratamientos combinados, especialmente con fármacos inhibidores de la vía de las MAP cinasas8,10,14,54,61 o, incluso, inhibiendo simultáneamente dos puntos de la vía PI3K/AKT63 (ver apartado: Tratamiento combinado). Sin embargo, existe un trabajo, desarrollado en un modelo murino de melanoma, en el que se sugiere que la inhibición de la vía PI3K/AKT podría inducir una inmunosupresión del huésped que acabaría favoreciendo el crecimiento del tumor64.
La inhibición mediante fármacos de otras moléculas implicadas en otras vías relacionadas con la proliferación/supervivencia, como CDK4 o ciclina D1, frecuentemente amplificadas en el melanoma, aún no se ha podido conseguir de forma adecuada60.
Inhibidores de moléculas de acción pleiotrópicaDenominamos así a las moléculas que actúan sobre múltiples funciones celulares distintas. De todos ellos, comentaremos los inhibidores del proteasoma y los inhibidores de deacetilasas de histonas. Estos inhibidores afectan a funciones importantes para el desarrollo y progreso de las neoplasias favoreciendo la parada del ciclo celular, la apoptosis, la disminución de las capacidades de invasión y migración celulares, la generación de especies reactivas de oxígeno, la inhibición de la angiogénesis y la autofagia.
Inhibidores del proteasomaEl proteasoma es un complejo enzimático encargado de la degradación de las proteínas de origen intracelular, que incluyen a proteínas implicadas en diversas funciones fundamentales de la célula. Se ha comprobado que la inhibición del proteasoma provoca apoptosis celular, tanto en células normales como en células malignas, pero de modo más intenso en estas últimas. Además, la inhibición del proteasoma sensibiliza indirectamente a la muerte celular inducida por otros mecanismos proapoptóticos como la quimioterapia o la radioterapia65. Los inhibidores del proteasoma, concretamente el denominado bortezomib (PS-341) se utilizan en el tratamiento del mieloma múltiple y de otras neoplasias hematológicas. Sin embargo, no parecen ser tan efectivos en tumores sólidos. Se ha demostrado la capacidad de los inhibidores del proteasoma para inducir apoptosis e inhibir el crecimiento celular en modelos experimentales in vitro o in vivo de melanoma66–68. No obstante, el primer ensayo clínico en melanoma diseminado en el que se empleó bortezomib en monoterapia no dio resultados satisfactorios69. Tampoco parecen muy esperanzadores los resultados de un ensayo clínico fase I que combina bortezomib con temozolamida70. A pesar de ello, se sigue trabajando en las posibilidades de tratamientos combinados con otras terapias71–79 y en los inhibidores del proteasoma de nueva generación80.
Inhibidores de deacetilasas de histonasUna de las características de las células neoplásicas es la alteración de la regulación de la expresión génica que se modula mediante mecanismos epigenéticos. Los mecanismos epigenéticos son mecanismos heredables que permiten ampliar la variabilidad de expresión del genoma de un individuo al regular la expresión de sus genes sin necesidad de modificar la secuencia de ADN de los mismos. Unos de los mecanismos epigenéticos mejor conocidos es el silenciamiento de genes mediante la metilación del promotor. La acetilación de histonas es otro de estos mecanismos. Las deacetilasas de histonas son un grupo de enzimas que inducen el silenciamiento de genes mediante la eliminación de grupos acetilo de las histonas. Al actuar sobre genes clave para la célula, su inhibición conduce a múltiples alteraciones celulares. El inhibidor de deacetilasas de histonas denominado vorinostat (SAHA) ya ha sido aprobado para el tratamiento de los linfomas cutáneos de células T81,82. En el melanoma se han obtenido resultados esperanzadores mediante el uso de distintos inhibidores de deacetilasas de histonas in vitro o en modelos animales83–87, especialmente en combinación con otros tratamientos como citostáticos, radioterapia, retinoides, inmunoterapia u otros fármacos frente a dianas terapéuticas88–92. No obstante, los resultados de los primeros ensayos clínicos fase I o II, ya sea en monoterapia o mediante terapia combinatoria, son discretos93–96.
Dianas moleculares que vehiculizan los mecanismos de invasión y metástasis: terapia antimoléculas de adhesiónLa gran capacidad de invasión y migración de las células de melanoma se debe, entre otros motivos, a la expresión de un patrón anómalo de moléculas de adhesión que las diferencia de los melanocitos normales. Ello les facilita, por ejemplo, desprenderse de los queratinocitos epidérmicos (pérdida de caderina E), adherirse a los fibroblastos y al endotelio vascular (sobreexpresión de caderina N), pasar de la fase de crecimiento radial a crecimiento vertical y metastatizar (sobreexpresión de MCAM/MUC18) o adherirse a las proteínas de la matriz extracelular y secretar metaloproteinasas para degradarlas (sobreexpresión de la integrina αvβ3)97,98. Además, la inhibición de algunos tipos de moléculas de adhesión, como las integrinas, puede también inhibir la proliferación e inducir la apoptosis de las células neoplásicas y ejercer un efecto antiangiogénico99,100.
Una de las principales dianas contra la que se ha actuado ha sido la integrina αvβ3 frente a la que se han producido anticuerpos monoclonales como etaracizumab (MEDI-522). En estudios preclínicos se había demostrado su actividad antitumoral como tratamiento único o en combinación con inhibidores de MCAM o de caderina N101. Sin embargo, su utilización en ensayos clínicos fase I y II, en monoterapia o en combinación con citostáticos, no ha ofrecido resultados esperanzadores102. Tampoco ha resultado convincente el tratamiento mediante inhibidores de la caderina N (ADH-1)103. En conjunto, la terapia dirigida a inhibir moléculas de adhesión no parece ser, por ahora, muy prometedora para el tratamiento del melanoma metastásico97,98.
Dianas terapéuticas localizadas en estructuras ajenas a las células neoplásicasDianas terapéuticas localizadas en el entorno tumoral: fármacos antiangiogénicosEl melanoma es un tumor asociado a una intensa angiogénesis que permite y mantiene el crecimiento tumoral y facilita su actividad metastatizante. Estos hechos sugieren que la terapia antiangiogénica es probablemente otra de las estrategias terapéuticas que puede ser útil en el melanoma104. Uno de los fármacos antiangiogénicos que más se ha ensayado clínicamente es bevacizumab, un anticuerpo monoclonal contra VEGF. Se ha empleado en el melanoma como único fármaco, sin mucho éxito, o asociado a citostáticos (carboplatino + placlitaxel o temozolamida) o a interferón-alfa. En uno de los últimos ensayos fase II presentado en el congreso ECCO-ESMO de 2009 sobre 214 pacientes105, en los que un grupo recibió carboplatino, placlitaxel y bevacizumab y otro grupo únicamente carboplatino, placlitaxel y placebo, se observó un mayor porcentaje de respuestas, de supervivencia global y de supervivencia a un año en el grupo que recibió bevacizumab, pero ninguna de estas diferencias era estadísticamente significativa8,17,61,100.
Otros de los fármacos antiangiogénicos propuestos, aunque de momento sin efectos clínicos claramente remarcables, son inhibidores multicinasa de los diferentes receptores de VEGF (VEGFR-1, 2 y -3) (sunitinib, sorafenib, semaxinib, axatinib etc.), derivados de la talidomida o fármacos antimoléculas de adhesión8,17,61,100,104,106.
Dianas terapéuticas facilitadoras de los mecanismos de escape tumoral: anticuerpos anti CTLA-4 y otras formas de inmunoterapia frente a dianas molecularesEl melanoma es un tumor enormemente inmunogénico que desencadena con facilidad una respuesta del sistema inmune contra las células tumorales. Sin embargo, las células neoplásicas utilizan una serie de estrategias para burlar esta respuesta. Una de las formas de luchar contra estos mecanismos es inhibir un receptor de los linfocitos T, denominado CTLA-4, inductor de inmunotolerancia. CTLA-4 compite con la molécula CD28 linfocitaria en su unión con la molécula B7 de las células presentadoras de antígeno. La unión CTLA-4/B7, en lugar de estimular la respuesta inmune citotóxica, induce anergia. Por ello, la inhibición de CTLA-4 puede romper la inmunotolerancia frente al melanoma. Se han utilizado dos tipos de anticuerpos monoclonales anti CTLA-4 en pacientes con melanoma metastásico, ipilimumab y tremelimumab107. Ipilimumab es el que se ha desarrollado de una forma más rápida y con un mayor éxito clínico107–109, de forma que el tratamiento del melanoma metastásico mediante ipilimumab ha sido aprobado recientemente por la FDA (USA Food and Drug Administration).
La valoración inicial de las respuestas clínicas frente a este tipo de fármacos ha resultado problemática. Los ensayos con sustancias inmunomoduladoras, como los anticuerpos anti CTLA-4, plantean un problema referente a la evaluación de la respuesta al tratamiento ya que pueden inducir una respuesta bifásica caracterizada por un empeoramiento inicial, pero un beneficio posterior y duradero. Este tipo de respuesta no puede evaluarse de forma adecuada con los sistemas actuales empleados en oncología (criterios de la OMS o criterios RECIST). Por ello, se han redefinido unos nuevos criterios dirigidos a evaluar específicamente este tipo de respuesta, en los que tiene más valor la supervivencia global y la estabilización de la enfermedad que la tasa de respuesta inicial valorada con los métodos clásicos107,108.
El último ensayo fase III publicado109 incluyó 676 pacientes que se randomizaron en tres brazos. Un brazo (403 pacientes) recibió ipilimumab junto a la vacuna peptídica gp100, otro brazo (177 pacientes) solo ipilimumab y otro (136 pacientes), únicamente la vacuna. No se observaron diferencias significativas entre los dos brazos que recibieron ipilimumab. Sin embargo, la mediana de supervivencia global del brazo que recibió ipilimumab y la vacuna gp100 fue significativamente superior que la del brazo que recibió únicamente la vacuna (10 meses vs 6,4 meses; p<0,001). En conjunto, los resultados de los ensayos clínicos publicados, indican que, tal como ocurría con los inhibidores específicos de BRAF, una de las ventajas de ipilimumab es que induce respuestas en enfermos con metástasis de mal pronóstico, incluyendo metástasis del sistema nervioso central, y/o con concentraciones elevadas de LDH sérica. Otra de las ventajas, ésta inherente a este tipo de tratamiento, es que una vez alcanzada la respuesta (10% completa o parcial, 10-20% estabilización, 10% progresión inicial y respuesta clínica posterior), ésta tiende a mantenerse en el tiempo con seguimientos ya de hasta más de 5 años en algunos casos. Existe además la posibilidad de volver a tratar con un 50% de éxito a los enfermos en el caso de que recaigan meses o años después de haber suspendido el tratamiento107. Los principales efectos secundarios de los cuales un 10-15% son de grado 3 o superior, están relacionados con la inmunidad y consisten en dermatitis, colitis, hipofisitis, tiroiditis y hepatitis. Puede producirse perforación del colon, por lo que los pacientes con diarrea deben controlarse de forma estricta. Se han desarrollado algoritmos específicos que permiten manejar de modo más seguro la toxicidad peculiar de los anticuerpos anti CTLA-4. Uno de los aspectos en el que se está trabajando es el hallar marcadores predictivos que permitan seleccionar a aquellos enfermos que pueden beneficiarse del tratamiento. Ello es importante, tanto para insistir con el tratamiento en casos que presenten progresión inicial, como para evitar la toxicidad, nada despreciable, en pacientes potencialmente no respondedores107,110.
Por otro lado, se está ensayando la terapia combinatoria junto a citostáticos, a fármacos frente a otras dianas y evaluando su papel como terapia adyuvante del melanoma de alto riesgo107,110. Otras moléculas que podrían inhibirse con una estrategia similar y en las que ya se está trabajando son CD137, OX40 y PD12,110.
Tratamientos combinadosPor los resultados clínicos obtenidos hasta el momento, parece difícil que enfocando el tratamiento del melanoma metastásico hacia una única diana seamos capaces de conseguir una remisión estable de la enfermedad. Una explicación de la aparición de resistencias a los tratamientos empleados es que las células de melanoma desarrollen respuestas compensatorias o que existan ya previamente vías de señalización redundantes que se activen cuando algunas de las vías se interrumpan. Por lo tanto, una estrategia para evitar o combatir estos fenómenos consistiría en utilizar simultáneamente fármacos frente a más de una diana. Dos de las combinaciones más avaladas son la inhibición simultánea de dos moléculas de la vía de las MAP cinasas (especialmente BRAF y MEK, lo que constituiría una «inhibición vertical» de la vía de las MAP cinasas) o de una molécula de la vía de las MAP cinasas y de una molécula de la vía PI3K/AKT. También se ha propuesto recientemente la «inhibición vertical» de la vía PI3K/AKT actuando simultáneamente sobre PI3K y mTOR63. Sin embargo, existen muchas otras posibilidades que incluyen desde combinación con citostáticos convencionales, moléculas de acción pleiotrópica, distintas formas de inmunoterapia o cualquiera de los tratamientos frente a las dianas que hemos mencionado8,54,60,97,110,112.
Además de las comentadas, la lista de posibles dianas que se están investigando para el tratamiento del melanoma es muy larga (Hsp90, ERBB4, GNaQ, SSTRs, CDK2, iNOS, FGFR-1, APP, etc.)14,54,97,111 y seguramente seguirá aumentando. En conjunto, de todos los fármacos ensayados hasta el momento, los que parecen poseer mayor futuro son los inhibidores específicos de BRAF, en los melanomas con la mutación V600E de BRAF, los inhibidores de c-Kit, en aquellos melanomas lentiginosos de piel y mucosas con mutaciones de c-Kit y el anticuerpo monoclonal anti-CTLA4 capaz de romper la inmunotolerancia del sistema inmune frente al melanoma. Estas opciones significan un gran cambio respecto a lo que teníamos hasta hace pocos años, pero, no obstante, aún quedan muchos problemas por resolver. En primer lugar, es importante diseñar estrategias para evitar o para saber cómo combatir la aparición de resistencias frente a los inhibidores de BRAF y c-Kit. Por otro lado, también es indispensable identificar biomarcadores que permitan una selección de los pacientes potencialmente susceptibles a terapias del tipo de ipilimumab. Se deberían encontrar dianas contra las que se pudiera luchar de forma efectiva en aquellos pacientes con mutaciones distintas a BRAF/V600E o a c-Kit. Y se deberían estudiar las posibilidades reales de la terapia combinada. Todo esto pasa por aprovechar toda la información que se pueda extraer de los ensayos clínicos, que, obviamente deben estar diseñados con este fin (introducción de pacientes correctamente tipificados, recolección de muestras durante el ensayo para diversos estudios moleculares, etc.), y por seguir trabajando en estudios preclínicos. Por primera vez da la sensación de que se ha abierto un nuevo horizonte sobre las posibilidades de tratamiento del melanoma diseminado. Sin embargo solo se han dado los primeros pasos; el cambio real no ha hecho más que empezar20,60.
FinanciaciónEste trabajo ha sido subvencionado por las ayudas FIS-PI060832, 2009SGR794 y RD06/0020/1034, una beca predoctoral de la AECC, Catalunya contra el Càncer, Lleida para A.S. y un contrato postdoctoral Juan de la Cierva para A.Y.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Con posterioridad a la redacción de este manuscrito, en julio de 2011, la Agencia Europea del Medicamento (EMA) aprobó Ipilimumab (Yervoy®) para el tratamiento del melanoma avanzado (metastásico o irresecable) resistente a, al menos, un tratamiento previo (ya aprobado por la FDA en marzo del 2011). En agosto del 2011, la FDA aprobó PLX4032 o Vemurafenib (Zelboraf®) como tratamiento de primera línea del melanoma metastásico y/o irresecable quirúrgicamente.


