






En los últimos años se han producido importantes avances en la genética de las porfirias y, concretamente, en las porfirias eritropoyéticas —protoporfiria eritropoyética (PPE) y porfiria eritropoyética congénita (PEC)—, que han dado lugar a una nueva concepción de las mismas como enfermedades no monogénicas. Se han identificado mutaciones en nuevos genes como responsables o modificadores de la gravedad de la porfiria, permitiendo esclarecer las discrepancias geno-fenotípicas observadas entre pacientes portadores de las mismas mutaciones, así como identificar el defecto genético responsable de algunos casos de porfiria en los que los estudios moleculares del gen uroporfirinógeno sintetasa (UROS) en la PEC o del gen ferroquelatasa (FECH) en la PPE no identificaban ninguna mutación. La mejor caracterización y conocimiento de la genética de estas enfermedades permitirá establecer cuadros geno-fenotípicos concretos y realizar una clasificación molecular con implicaciones prácticas y pronósticas.
In recent years, important advances have been made in our understanding of the genetics of porphyrias, particularly with respect to erythropoietic protoporphyria (EPP) and congenital erythropoietic porphyria (CEP), 2 forms of erythropoietic porphyria no longer considered to be monogenic. The identification of mutations in genes not previously associated with these disorders as causative factors or modulators of severity has helped to explain the presence of genotypic and phenotypic differences between patients carrying the same mutations. These advances have also led to the identification of causative genetic defects in patients who, based on molecular studies, had no mutations in the uroporphyrinogen III synthase gene UROS (in CEP) or in the ferrochelatase gene FECH (in EPP). Better understanding and characterization of the genetics of porphyrias will allow us to determine genotypic and phenotypic correlations and improve the molecular classification of these diseases, which will have both practical and prognostic implications.
Las porfirias son un grupo de enfermedades metabólicas causadas por deficiencias en las enzimas de la vía de síntesis del grupo hem. En función de la enzima alterada se acumulan los distintos sustratos (precursores y porfirinas) que desencadenan las manifestaciones clínicas propias de cada porfiria (fig. 1). Hasta hace poco se habían descrito mutaciones en los 7 genes —aminolevulinato deshidratasa (ALAD), porfobilinógeno desaminasa (PBGD), uroporfirinógeno sintetasa (UROS), uroporfirinógeno decarboxilasa (UROD), coproporfirinógeno oxidasa (CPOX), protoporfirinógeno oxidasa (PPOX) y ferroquelatasa (FECH)— responsables de los 7 tipos de porfiria reconocidos. Sin embargo, en los últimos años se ha demostrado la participación de otros genes que codifican para proteínas que no forzosamente forman parte de la vía de biosíntesis del grupo hem como causantes de formas concretas de porfiria.
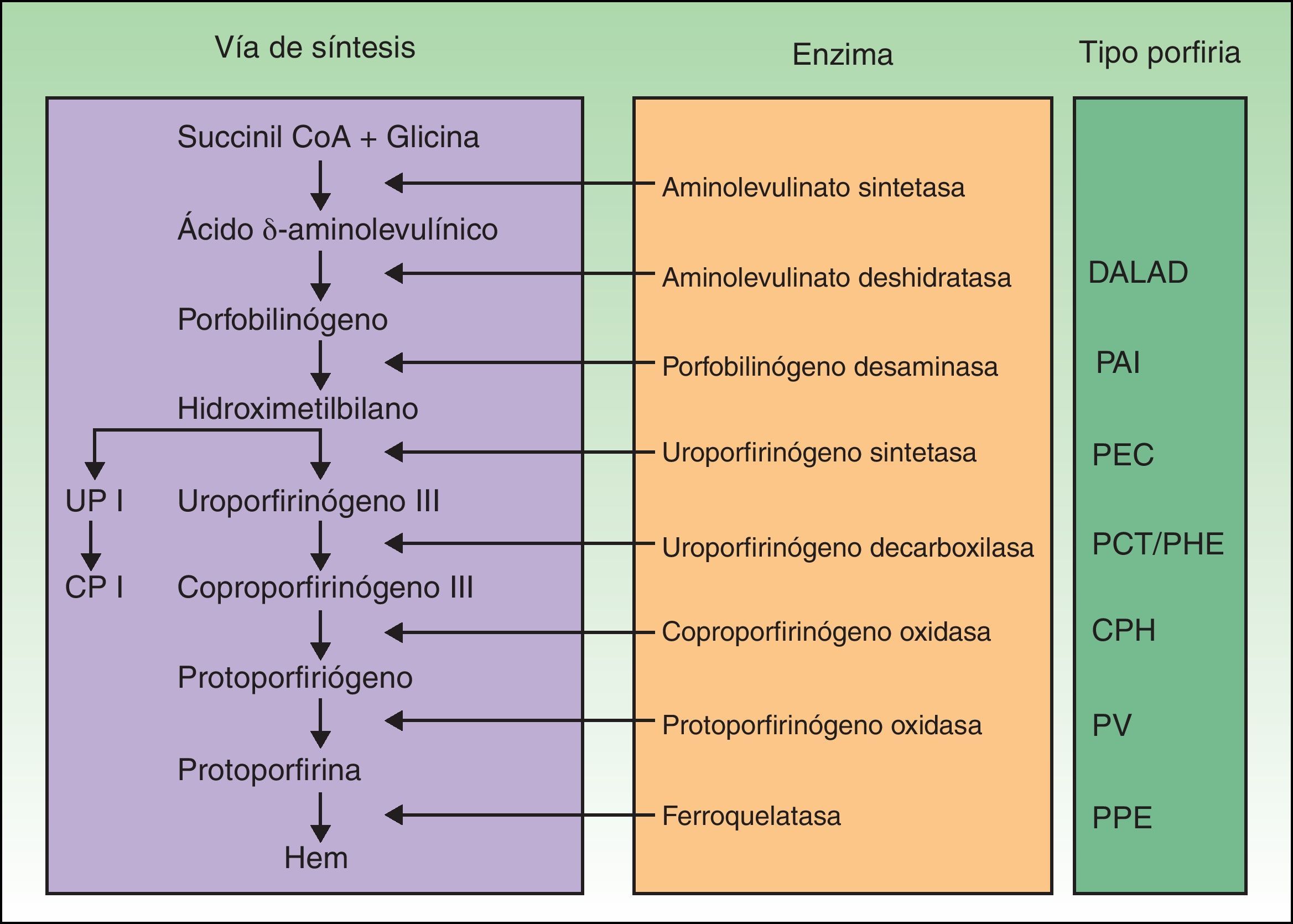
Clasificación de las porfirias. CP I: coproporfirinas isómero I; CPH: coproporfiria hereditaria; DALAD: porfiria por déficit de aminolevulinato deshidratasa; PAI: porfiria aguda intermitente; PCT: porfiria cutánea tarda; PEC: porfiria eritropoyética congénita; PHE: porfiria hepatoeritropoyética; PPE: protoporfiria eritropoyética; PV: porfiria variegata; UP I: uroporfirinas isómero I.
Los avances en el campo de la genética de las porfirias, los estudios bioquímicos y la identificación de rasgos clínicos concretos hacen que las porfirias sean un grupo de enfermedades en continua renovación. En este sentido, las mayores novedades se han producido en las porfirias eritropoyéticas: la protoporfiria eritropoyética (PPE) y la porfiria eritropoyética congénita (PEC). Por este motivo, la presente actualización se centrará en las últimas novedades de estas dos enfermedades, cuya manifestación clínica principal asienta en la piel y el sistema hematopoyético.
Protoporfiria eritropoyéticaLa protoporfiria eritropoyética (PPE) es una enfermedad hereditaria que se caracteriza por el desarrollo, ya en edades muy tempranas, de un cuadro de fotosensibilidad cutánea que perdura a lo largo de la vida del paciente. Se trata de un trastorno poco común, con prevalencias que varían de 1/143.000 en el Reino Unido a 1/75.000 en Holanda1,2. Sin embargo, se trata de la forma más frecuente de porfiria en la edad pediátrica.
La enfermedad se debe a deficiencias parciales en la actividad de la última enzima de la vía de síntesis del grupo hem, ferroquelatasa. Este déficit funcional es secundario a mutaciones en el gen FECH, localizado en el brazo largo del cromosoma 18 (concretamente en el locus 18q21.3)3, y hasta la fecha se han identificado más de 120 mutaciones distintas responsables de la enfermedad. La enzima ferroquelatasa cataliza la inserción del hierro en el interior del anillo de protoporfirina IX (PpIX), siendo la acumulación de esta última molécula en los eritrocitos, el plasma, la piel y el hígado la responsable del cuadro clínico.
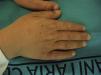
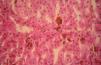
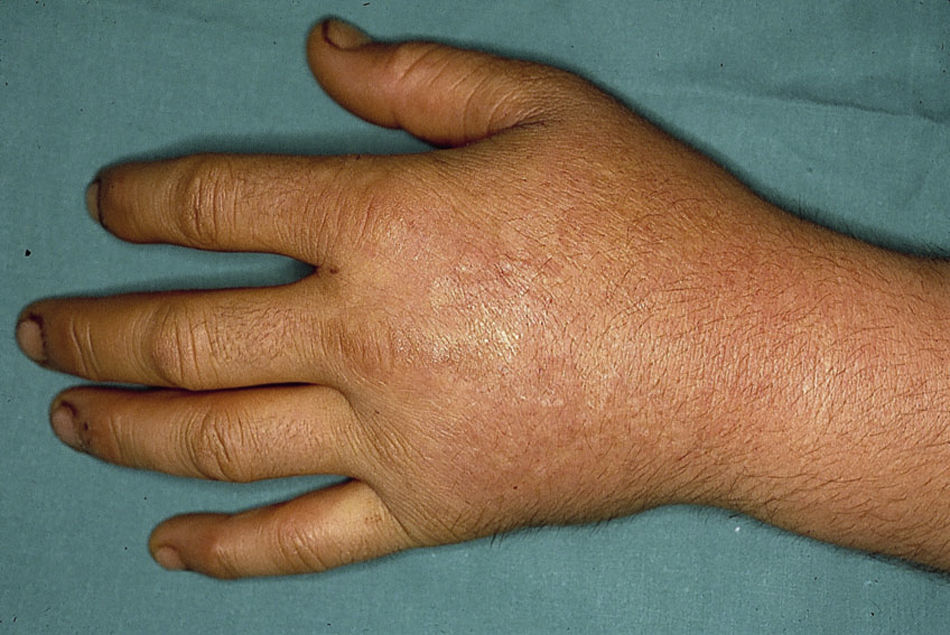
El cuadro clínico de la PPE es distinto al del resto de porfirias cutáneas. La PPE se manifiesta en forma de un cuadro agudo de fotosensibilidad con eritema, edema y sensación dolorosa de quemazón, muy similar a una quemadura solar, que aparece tras 1-30 minutos de exposición solar (fig. 2). Por este motivo, los pacientes sufren una importante limitación para la realización de actividades al aire libre que disminuye notablemente su calidad de vida1,4. La exposición solar repetida puede producir lesiones cutáneas crónicas en las zonas fotoexpuestas. Se trata de lesiones poco invalidantes, en forma de cicatrices puntiformes y «varioliformes» faciales, así como un engrosamiento «céreo» de la piel, particularmente de la región perioral y encima de las articulaciones metacarpofalángicas (fig. 3)1,5.
Debido a las características intrínsecas de la PpIX (hidrofobia y liposolubilidad), esta molécula tiene un aclaramiento hepático, eliminándose a través de la bilis y las heces. Un exceso de PpIX en los canalículos biliares puede producir una hepatopatía colestásica. En este sentido, hasta un 25% de los pacientes pueden padecer de colelitiasis por cálculos de porfirinas, y hasta un 5% pueden desarrollar una hepatopatía grave que conduce a la insuficiencia hepática, cuya única solución es el trasplante hepático6.
Casi la mitad de los pacientes presentan anemia, normalmente leve y asintomática, con parámetros de ferropenia (microcitosis e hipocromía) refractaria a los suplementos de hierro7.
Bioquímicamente la PPE se caracteriza por un aumento de la concentración de PpIX libre (no unida a Zn) en los hematíes y en el plasma. Por este motivo, el plasma emite fluorescencia con un pico máximo de emisión en torno a los 634nm. A diferencia de otras porfirias cutáneas, no se detectan porfirinas en orina, ya que la PpIX se elimina exclusivamente por las heces8.
En los últimos años se han producido importantes descubrimientos moleculares en la PPE, gracias a los cuales se ha establecido el mecanismo de transmisión hereditario de las formas dominantes, se han identificado subgrupos de pacientes con unas características genotípicas y fenotípicas concretas, y se ha demostrado la existencia de nuevas formas de la enfermedad debidas a mutaciones en genes distintos al que codifica la FECH. Ello ha permitido actualizar el abordaje de la PPE no como una enfermedad monogénica, sino como una enfermedad heterogénea en la que no siempre resulta sencillo establecer una correlación entre el genotipo y el fenotipo de la enfermedad. Los recientes conocimientos sobre la genética de la PPE han permitido establecer una clasificación basada en criterios moleculares, pero con evidente aplicación clínica, sobre todo en relación con el riesgo de desarrollar hepatopatía grave (fig. 4).
Forma autosómica dominante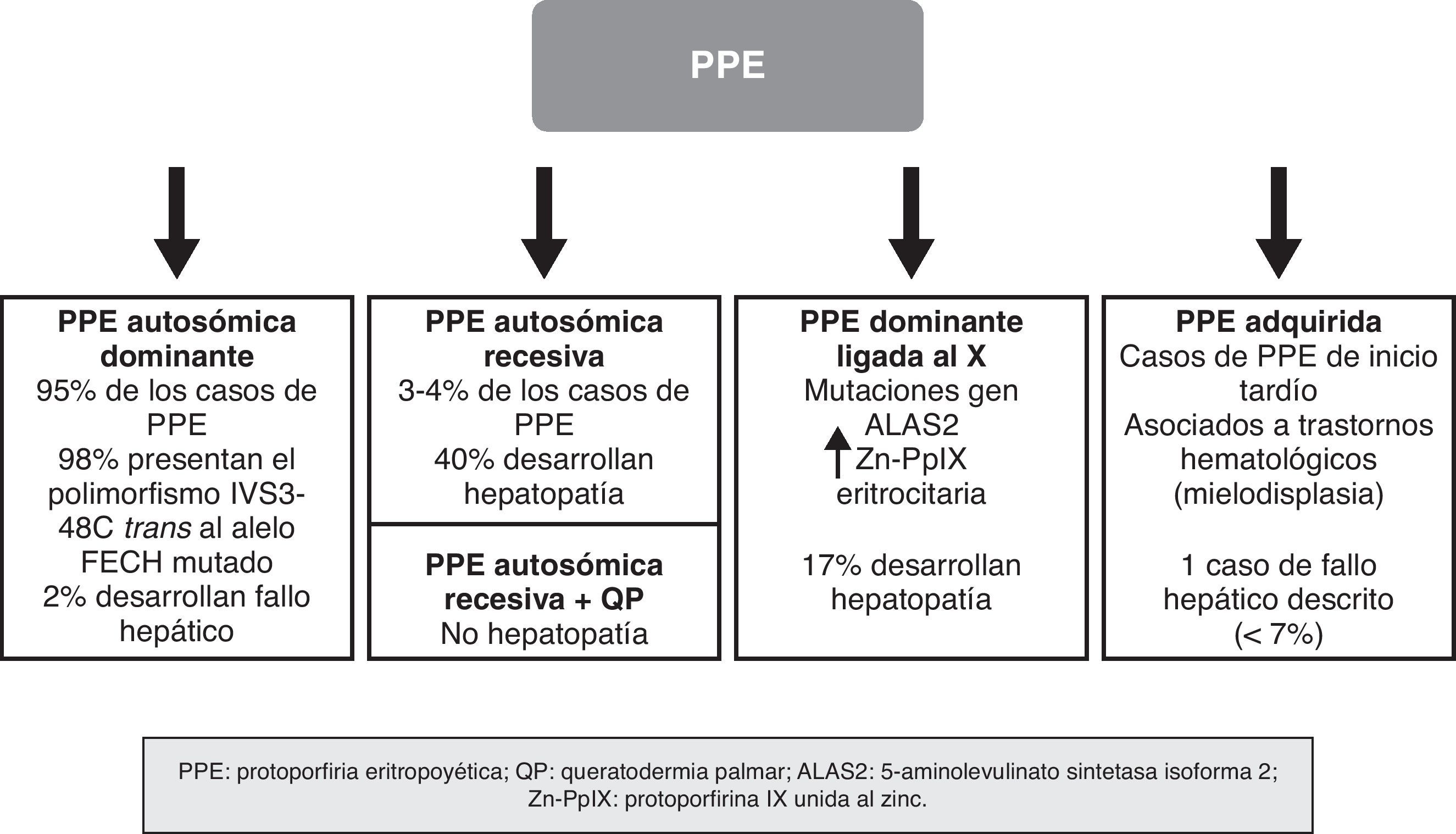
Se trata de la forma más común de PPE, representando casi el 95% de los casos. Se debe a la transmisión de una mutación en uno de los dos alelos del gen FECH que conlleva una alteración cuantitativa o cualitativa de la enzima ferroquelatasa. El grado de actividad enzimática residual varía entre los pacientes y los familiares portadores de la mutación, determinando un patrón de herencia autosómico dominante con penetrancia incompleta. Así, mientras los portadores de uno de los dos alelos mutado muestran una actividad enzimática residual en torno al 50%, los individuos sintomáticos muestran unos valores inferiores al 35% de la actividad normal9. Durante años se propuso que esta reducción adicional en la actividad enzimática podía estar en relación con la presencia de un polimorfismo común en el alelo no mutado, que llevaría a una expresión del gen por debajo de un umbral crítico necesario para la manifestación de la enfermedad10,11. En 2002 Gouya identificó un polimorfismo en el intrón 3 del gen FECH, IVS3-48C, como responsable de la baja expresión del gen FECH no mutado, presente en el 98% de los enfermos12. El cambio de un único nucleótido, IVS3-48T>C, produce un mayor porcentaje de ARNm aberrante que es rápidamente degradado, llevando a una disminución en la expresión de la proteína. Este hallazgo ha permitido entender el mecanismo por el que solo algunos de los portadores de un gen mutado desarrollan la enfermedad. De este modo, para que se manifieste la PPE es necesario que se cohereden el alelo mutado y el alelo de baja expresión (IVS3-48C). La prevalencia de este último varía ampliamente entre las distintas poblaciones13. Así, por ejemplo, se ha descrito una prevalencia del 43% en Japón, muy superior a la encontrada en la población de África occidental (<1%)14,15. Sin embargo, parece ser bastante homogénea entre la población europea, detectándose una frecuencia poblacional del 13, 11 y 10,5% en el Reino Unido, Francia y España, respectivamente12,16,17.
Hasta la fecha no se ha podido establecer una relación entre mutaciones específicas del gen FECH y la concentración de PpIX, la edad de inicio de la enfermedad y la severidad del cuadro cutáneo. No obstante, parece existir una correlación genotipo-fenotipo significativa entre portadores de mutaciones null allele (que producen una pérdida total de la función enzimática) y la afectación hepática grave18,19.
Forma autosómica recesivaAproximadamente el 4% de los pacientes con PPE son portadores de verdaderas mutaciones en ambos alelos, constituyendo la forma recesiva de la enfermedad. Aunque los casos reportados son escasos —alrededor de 20 pacientes— esta es la forma más frecuente de PPE en aquellos individuos no portadores del alelo de baja expresión16.
Clínicamente estos pacientes presentan las mismas características dermatológicas que los casos heterocigotos. Sin embargo, el riesgo de desarrollar hepatopatía grave y fallo hepático que requiera de trasplante es mucho mayor, siendo por ello aconsejable un seguimiento estricto de la función hepática y de las concentraciones de protoporfirinas en plasma y heces20 (fig. 4).
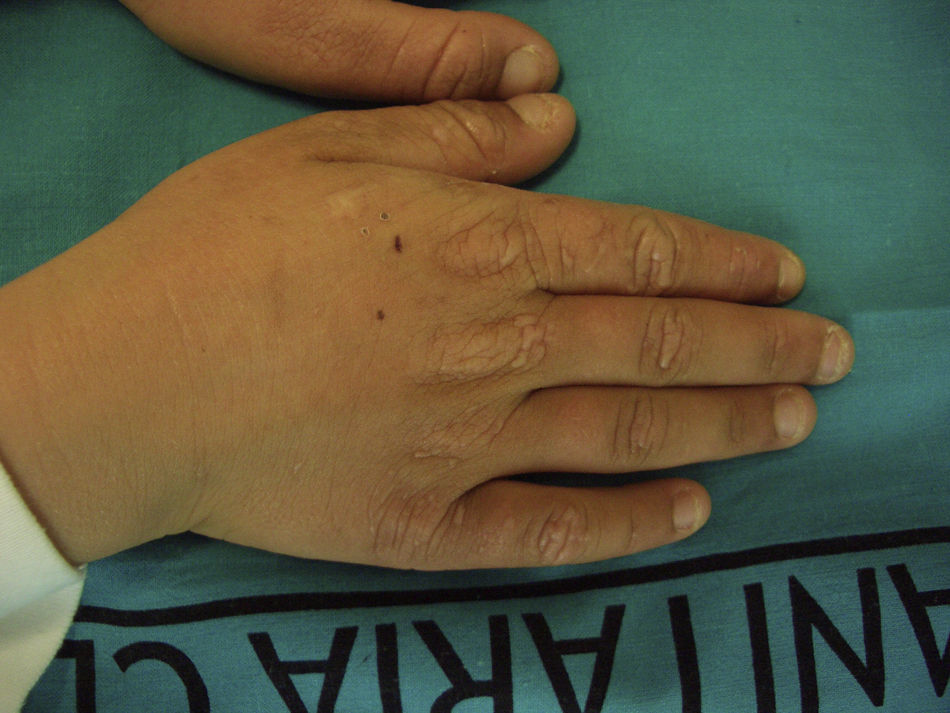
Recientemente Holme et al.21 han descrito un subgrupo de 9 pacientes con PPE recesiva (PPEr) afectos de una peculiar queratodermia palmar. Todos los pacientes presentaron la queratodermia desde la primera infancia y en 7 de ellos se desarrolló antes del diagnóstico de PPE. Todos mostraron una queratodermia bien delimitada, sin borde eritematoso y mínimamente transgrediens. El grado de hiperqueratosis fue muy variable, pero en todos ellos empeoraba en relación con la exposición solar, confiriéndole un carácter estacional. Además, todos ellos presentaban un engrosamiento de la piel de los nudillos, signo presente en el 35% de los pacientes con PPE. Bioquímicamente presentaron una menor concentración eritrocitaria de PpIX que los pacientes sin queratodermia. Casi todos los pacientes eran portadores de mutaciones missense (causantes de cambios puntuales de aminoácidos que llevan a la formación de una proteína alterada, pero que mantiene cierto grado de funcionalidad), aunque no se pudo establecer ninguna relación genotipo-fenotipo, pues algunas de estas mutaciones habían sido ya descritas en pacientes sin queratodermia. No obstante, el dato más significativo fue la ausencia de afectación hepática observada en todos los pacientes con queratodermia palmar, hecho que contrasta con el alto porcentaje de afectación hepática grave —de hasta un 42%— en el grupo de PPE recesiva sin queratodermia.
Así pues, la presencia de queratodermia palmar en edades muy tempranas podría considerarse un signo clínico específico de PPE recesiva, definiendo un subgrupo con poco riesgo de desarrollar hepatopatía.
Forma dominante ligada al cromosoma XWhatley et al.22 demostraron en el año 2008 un nuevo tipo de PPE en pacientes sin mutaciones en el gen FECH. Aproximadamente en un 7% de las familias con PPE estudiadas a nivel molecular no se encuentran mutaciones en el gen de la ferroquelatasa. Dentro de este subgrupo se han identificado 8 familias con individuos afectos de una PPE clínicamente idéntica a la de los pacientes con la forma autosómica dominante (PPEd). Sin embargo, mostraban unas características diferenciales, como son: a) una concentración eritrocitaria de PpIX mayor que los pacientes con PPEd; b) una proporción de Zn-PpIX mucho mayor que la forma de PPEd; y c) un patrón de herencia dominante ligado al cromosoma X. Este hallazgo motivó el estudio de los genes localizados en el cromosoma X que codifican la síntesis de proteínas importantes en la vía de biosíntesis del grupo hem, demostrándose que ciertas mutaciones en el gen de la ALAS2 eran responsables de esta nueva forma de PPE.
ALAS2 es la isoforma específica eritroide del gen 5-aminolevulinato sintetasa (ALAS), indispensable para la síntesis de la hemoglobina en las células eritroides y, hasta la fecha, solo se habían descrito mutaciones causantes de anemias sideroblásticas hereditarias. En el estudio de Whatley22 se identificaron 2 deleciones distintas (c.1706-1709 delAGTG y c.1699-1700 delAT) en el exón 11 del gen ALAS2, responsables de alteraciones en los aminoácidos 19-20 del extremo C-terminal de la enzima. Estas alteraciones conducen a un cambio estructural en la proteína que llevan a un aumento de función de la misma. Esta es la primera vez que se identifica una mutación causante de un incremento en la funcionalidad de una de las enzimas de la biosíntesis del hem. El aumento de funcionalidad de la enzima ALAS2, la enzima limitante de la vía metabólica en condiciones normales, hace de la ferroquelatasa la nueva enzima limitante, produciéndose una mayor acumulación de protoporfirina ligada a cinc.
Es importante destacar el alto porcentaje de pacientes que presentaron enfermedad hepática (17%), sugiriendo que la PPE ligada al cromosoma X (PPELX) conlleva un mayor riesgo de afectación hepática grave, de forma análoga a la variante autosómica recesiva.
Así pues, parece que las alteraciones de la región C-terminal de la ALAS2 conducen a la producción de protoporfirinas en exceso suficiente para causar fotosensibilidad y daño hepático, aunque la actividad de la ferroquelatasa sea completamente normal.
Formas adquiridas (de inicio tardío)La PPE adquirida, con inicio en la edad adulta (mayores de 18 años), ha sido descrita en relación con neoplasias hematológicas23. A pesar de que se trata de una entidad muy poco frecuente debemos tenerla en cuenta en aquellos casos de fotosensibilidad aguda grave que se inician en un adulto.
Se han publicado menos de 15 casos, principalmente asociados con síndromes de mielodisplasia, principalmente del subtipo anemia sideroblástica24. Los pacientes con mielodisplasia presentan una inestabilidad genética que conduce a la formación de anomalías cromosómicas complejas en el clon de células mielodisplásicas. Se ha postulado que el desarrollo de la PPE en estos casos podría ser el resultado de la expansión de un clon de células hematopoyéticas conteniendo solo un alelo del gen FECH, como consecuencia de deleciones de una de las dos copias del cromosoma 1824,25.
Aunque la prevalencia de afectación hepática asociada a esta forma de PPE es desconocida, al menos se ha descrito un caso de enfermedad hepática fatal26.
Tratamiento de la porfiria eritropoyéticaLa principal medida terapéutica en la PPE sigue siendo evitar la fotoexposición, incluso a través de los cristales de las ventanas. La administración oral de dosis altas de betacarotenos (30mg/kg de peso y día) y las distintas modalidades de fototerapia que se realizan suelen mejorar la tolerancia a la luz27–29. En este sentido, y más últimamente, parece que el empleo de fármacos análogos de la hormona estimulante de melanocitos (MSH [melanocyte stimulating hormone]) que también actúan como agonistas del receptor de melanocortina, podrían ser útiles, pues promueven la síntesis de melanina, aumentando la pigmentación cutánea y la tolerancia a la luz natural30.
Es controvertida la utilidad de la administración oral de hierro con el fin de corregir la anemia que presentan muchos pacientes con PPE, ya que en algunos casos puede empeorar la fotosensibilidad cutánea31. Sin embargo, en casos de anemia grave puede ser necesaria la administración endovenosa de hierro o, incluso, la transfusión.
La colestiramina (4-16g/d) puede usarse para aumentar la eliminación de protoporfirinas por vía biliar, reduciendo sus niveles plasmáticos y disminuyendo el riesgo de hepatopatía32. Los ácidos ursodesoxicólico y quenodesoxicólico se han utilizado también con este objetivo33. Sin embargo, la principal complicación de la PPE sigue siendo la hepatopatía colestásica por depósito de protoporfirinas, que puede conducir a un fallo hepático, agudo e irreversible, tributario de trasplante hepático (fig. 5). Desde la realización del primer trasplante hepático en un paciente con PPE en 197934 se han comunicado más de 40 pacientes trasplantados, con una supervivencia global del 77% al primer año y del 66% a los 5 años35. Sin embargo, deben tenerse en cuenta 3 grandes complicaciones: a) las reacciones fototóxicas inducidas por las luces de quirófano, que pueden evitarse con filtros amarillos que bloquean las radiaciones por debajo de los 460nm36;b) la aparición de una neuropatía motora postoperatoria en pacientes con niveles plasmáticos de PpIX muy elevados37; yc) la recurrencia de la hepatopatía por protoporfiria en el hígado trasplantado, que puede prevenirse con el trasplante hematopoyético35,38. Por ello, en caso de afectación hepática grave y progresiva debe considerarse la posibilidad de un trasplante secuencial hepático y de médula ósea o, idealmente, la realización de un trasplante de médula ósea antes de llegar a la fase de fallo hepático35,39.
Porfiria eritropoyética congénita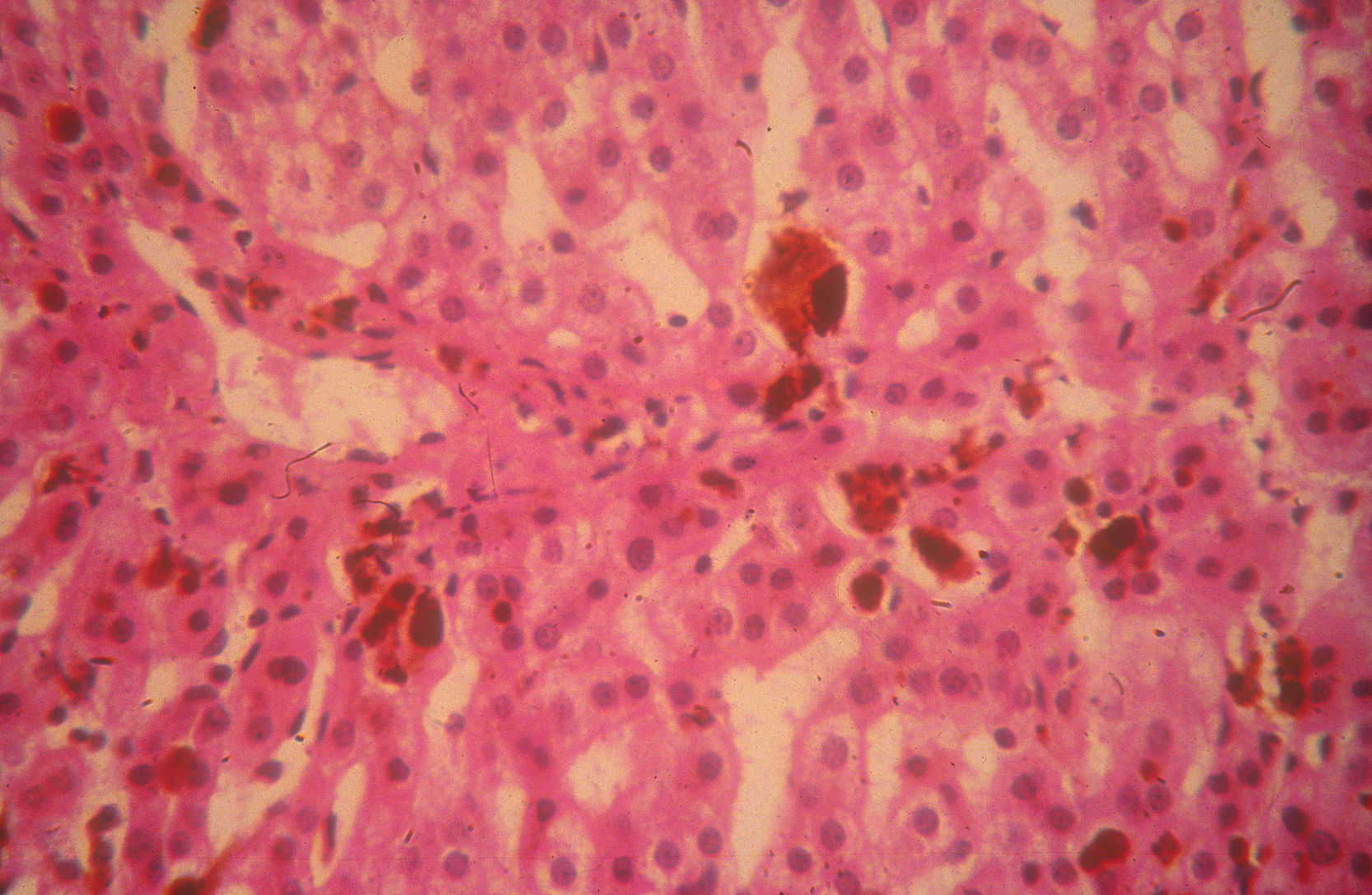
La PEC o enfermedad de Günther se hereda de forma autosómica recesiva. Es una enfermedad realmente infrecuente, con aproximadamente 150 casos descritos en la literatura40. Se debe a una deficiencia en la actividad de la enzima uroporfirinógeno-III sintetasa (UROS), codificada por el gen UROS que se localiza en el cromosoma 10q25.3→q26.341.
Las manifestaciones clínicas de la PEC son heterogéneas y muy variables. Pueden ir desde la aparición de un hydrops faetalis, que ocurre como consecuencia de una anemia hemolítica grave intraútero, hasta formas muy leves con apenas afectación cutánea en adultos (fig. 6). La emisión de orinas oscuras (rojizas) en un neonato puede ser la primera clave para el diagnóstico, aunque también puede verse en otras formas de porfiria (porfiria hepatoeritropoyética, formas recesivas de porfiria aguda intermitente, etc.)42.
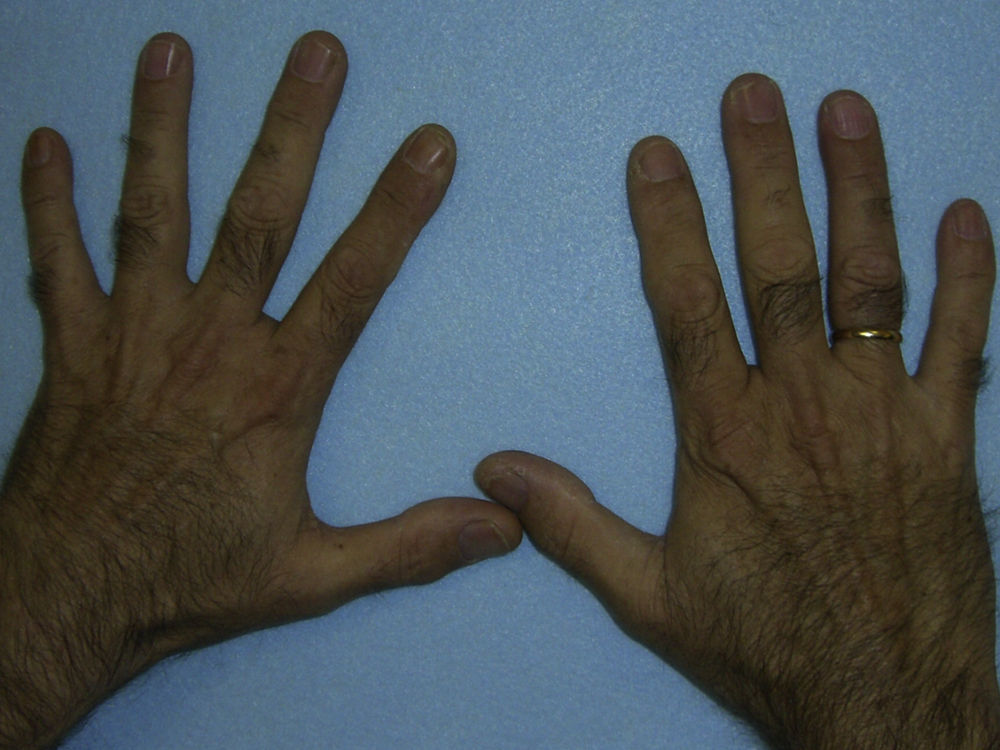
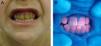
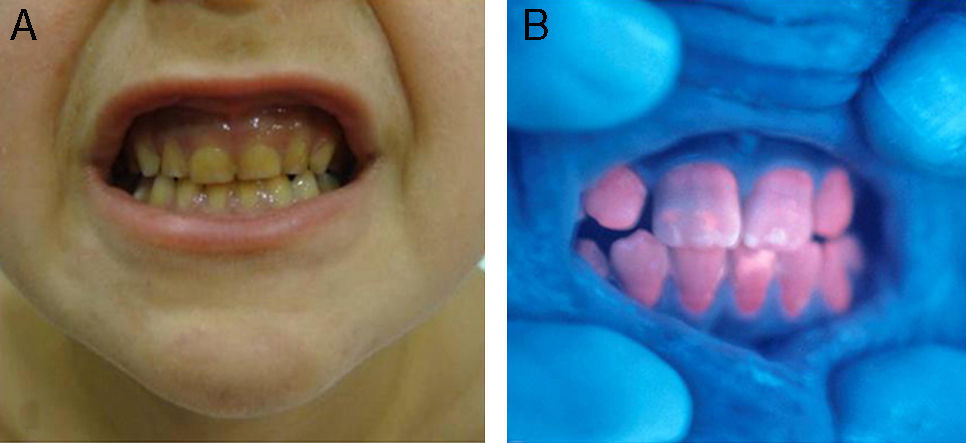
En general, los pacientes con PEC muestran una importante fotosensibilidad cutánea ya desde la primera infancia. La aparición recurrente de ampollas y la sobreinfección secundaria llevan a la aparición de cicatrices, mutilaciones y deformidades. La hipertricosis facial y generalizada es a menudo importante. Los depósitos de porfirinas en la córnea y en los dientes en formación pueden producir defectos visuales y eritrodoncia respectivamente (fig. 7).
La presencia de anemia hemolítica es muy frecuente. Los pacientes suelen mostrar anisocitosis, poiquilocitosis, reticulocitosis, ausencia de haptoglobina, hiperbilirrubinemia y aumento del urobilinógeno fecal. El hiperesplenismo secundario que se produce por eritropoyesis ineficaz contribuye a la anemia y puede también producir leucopenia y trombocitopenia. La anemia puede ser tan grave que se requieran transfusiones periódicas.
La PEC es la única porfiria que produce porfirinas de isómero I (uroporfirina I y coproporfirina I) en exceso y que pueden ser detectadas en orina o heces, respectivamente. Los hematíes contienen grandes cantidades de uroporfirina I y, en menor cantidad, coproporfirina I y protoporfirina-Zn (unida a cinc).
Se han descrito 43 mutaciones distintas del gen UROS43. La gran mayoría son mutaciones missense. La mutación p.C73R en el exón 4 es la mutación más común, con una frecuencia alélica del 30%, y en su forma homocigota se asocia a un fenotipo grave. Sin embargo, la mayoría de las mutaciones restantes han sido halladas en una o unas pocas familias de pacientes con PEC. Además, aunque de forma general se acepta que el grado de actividad residual de la enzima UROS en vivo determina el grado de afectación de la enfermedad, se han descrito pacientes portadores de las mismas mutaciones en el gen UROS con un fenotipo muy distinto44. Todo ello hace difícil poder establecer una correlación genotipo-fenotipo en la PEC y refuerza la hipótesis de la implicación de otros genes, distintos del gen UROS, que actuarían como genes modificadores de la enfermedad. Dos grandes descubrimientos recientes dejan patente esta posibilidad. El primero, demostrado por Phillips et al.45, fue la descripción del primer caso de PEC debido a una mutación en el factor de transcripción eritroide específico, GATA binding protein 1 (GATA-1), localizado en el cromosoma X (Xp11.23). Este factor de transcripción es vital para garantizar funciones como una eritropoyesis normal, una expresión adecuada de los genes de las globinas y un correcto desarrollo de los megacariocitos. Pero, además, actúa regulando la expresión del gen UROS en eritrocitos en desarrollo. Así pues, mutaciones en GATA-1 pueden producir PEC en ausencia de mutaciones en el gen UROS y, probablemente, actúe como otro gen modificador en algunos pacientes con mutaciones en UROS. El segundo hecho relevante, llevado acabo por To-Figueras et al.46, pone de manifiesto que mutaciones en el gen ALAS2 que producen un aumento de función de la enzima (gain-of-function mutations), tal y como se ha descrito en la PPELX, pueden actuar como factor agravante y producir un cuadro bioquímica y clínicamente más grave en pacientes con defectos en la UROS. Así pues, mutaciones en la región C-terminal del gen ALAS2 que llevan a una actividad enzimática incrementada, junto con mutaciones en el gen UROS que producen una disminución de la cinética enzimática a este nivel, conducen a una mayor acumulación de porfirinas de isómero I y, consecuentemente, a una clínica más grave.
Finalmente, cabe recordar la existencia de casos adquiridos de PEC, presentándose en la edad adulta. Se han publicado alrededor de 15 casos de PEC con comienzo en mayores de 18 años, normalmente mostrando una clínica más leve, y frecuentemente asociados con trombocitopenia y mielodisplasia, de forma análoga a la PPE adquirida47,48.
Tratamiento de la porfiria eritropoyética congénitaLos tres pilares básicos en el manejo terapéutico de los pacientes con PEC son: a) evitar la fotoexposición; b) un cuidado meticuloso de las heridas cutáneas; y c) transfusiones sanguíneas y otras medidas de soporte hematológico.
Otras medidas terapéuticas sintomáticas incluyen: la hidroxiurea49 para reducir la síntesis de porfirinas en la médula ósea, la esplenectomía para reducir las necesidades de transfusiones en pacientes con hiperesplenismo y la administración oral de carbón activado para facilitar la excreción fecal de porfirinas. Sin embargo, muchas de estas medidas no pueden considerarse como una solución a largo plazo, y en los casos más graves (pacientes transfusión-dependientes) el trasplante hematopoyético es el tratamiento de elección50. Se han publicado cerca de 15 pacientes con PEC que han recibido un trasplante alogénico de médula ósea, la mayoría de los casos con curación clínica y bioquímica de la enfermedad51. Aunque todavía no se dispone de la terapia génica como una opción real para la curación de esta porfiria, se han logrado avances experimentales notables que hacen prever su utilidad a medio plazo52.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


