







In recent years, important advances have been made in our understanding of the genetics of porphyrias, particularly with respect to erythropoietic protoporphyria (EPP) and congenital erythropoietic porphyria (CEP), 2 forms of erythropoietic porphyria no longer considered to be monogenic. The identification of mutations in genes not previously associated with these disorders as causative factors or modulators of severity has helped to explain the presence of genotypic and phenotypic differences between patients carrying the same mutations. These advances have also led to the identification of causative genetic defects in patients who, based on molecular studies, had no mutations in the uroporphyrinogen III synthase gene UROS (in CEP) or in the ferrochelatase gene FECH (in EPP). Better understanding and characterization of the genetics of porphyrias will allow us to determine genotypic and phenotypic correlations and improve the molecular classification of these diseases, which will have both practical and prognostic implications.
En los últimos años se han producido importantes avances en la genética de las porfirias y, concretamente, en las porfirias eritropoyéticas – protoporfiria eritropoyética (PPE) y porfiria eritropoyética congénita (PEC) -, que han dado lugar a una nueva concepción de las mismas como enfermedades no monogénicas. Se han identificado mutaciones en nuevos genes como responsables o modificadores de la gravedad de la porfiria, permitiendo esclarecer las discrepancias geno-fenotípicas observadas entre pacientes portadores de las mismas mutaciones, así como identificar el defecto genético responsable de algunos casos de porfiria en los que los estudios moleculares del gen uroporfirinógeno sintetasa (UROS) en la PEC o del gen ferroquelatasa (FECH) en la PPE, no identificaban ninguna mutación. La mejor caracterización y conocimiento de la genética de estas enfermedades permitirá establecer cuadros geno-fenotípicos concretos y realizar una clasificación molecular con implicaciones prácticas y pronósticas.
Porphyrias are a group of metabolic diseases caused by deficiencies in the enzymes that participate in the heme synthesis pathway. Deposits of porphyrins or their precursors (depending on the substrate of the affected enzyme) trigger the clinical manifestations characteristic of each porphyria (Fig. 1). Until recently, mutations had been described in the 7 genes (aminolevulinate dehydratase [ALAD], porphobilinogen deaminase [PBGD], uroporphyrinogen synthase [UROS], uroporphyrinogen decarboxylase [UROD], coproporphyrinogen oxidase [CPOX], protoporphyrinogen oxidase [PPOX], and ferrochelatase [FECH]) responsible for the 7 recognized types of porphyria. However, in the past few years, other genes encoding proteins that are not essential for the heme synthesis pathway have been identified as causing other specific forms of porphyria.
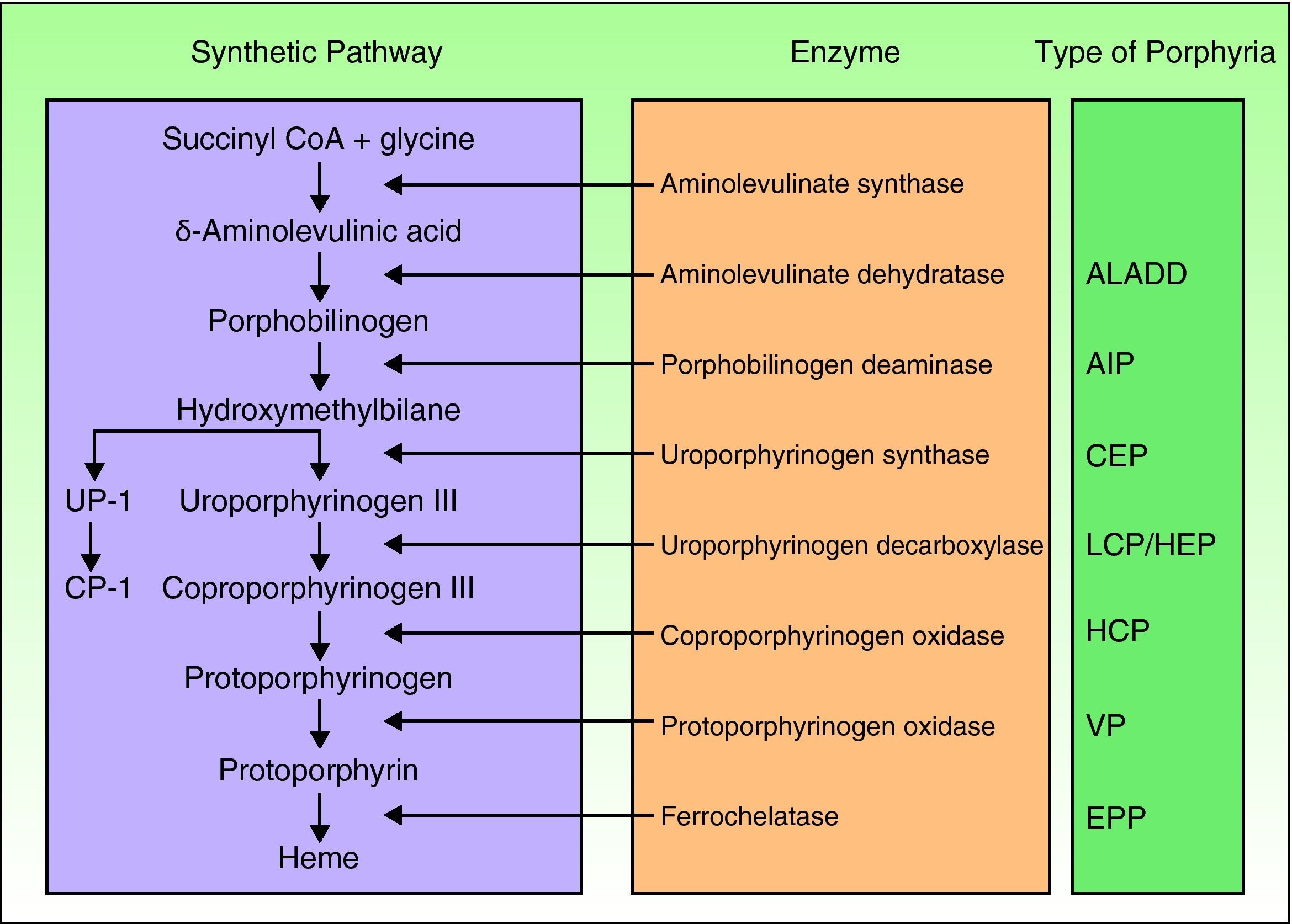
Classification of the porphyrias. CoA indicates coenzyme A; ALADD, porphyria arising from aminolevulinate dehydratase deficiency; AIP, acute intermittent porphyria; CEP, congenital erythropoietic porphyria; UP I, uroporphyrin I; LCP, late cutaneous porphyria; HEP, hepatoerythropoietic porphyria; CP I, coporphyrin isomer I; HCP, hereditary coproporphyria; VP, variegate porphyria; EPP: erythropoietic protoporphyria.
The porphyrias are a group of diseases in state of constant flux given the continuous advances in the field of porphyria genetics. These advances mainly involve the erythropoietic porphyrias: erythropoietic protoporphyria (EPP) and congenital erythropoietic porphyria (CEP). Thus, in this update, the focus will be on recent developments in these 2 diseases, whose effects are mainly manifest in the skin and hematopoietic system.
Erythropoietic ProtoporphyriaEPP is a hereditary disease characterized by cutaneous photosensitivity which develops at an early age and lasts throughout the life of the patient. It is an uncommon disorder, with a prevalence that varies between 1/143 000 in the United Kingdom and 1/75 000 in The Netherlands.1,2 It is, however, the most common porphyria in children.1,2
The disease is caused by partial deficiency in the activity of ferrochelatase, the last enzyme in the heme synthesis pathway. This functional deficiency is caused by mutations in the FECH gene, located on the long arm of chromosome 18 (specifically on locus 18q21.3).3 To date, more than 120 different mutations have been identified as responsible for the disease. The ferrochelatase enzyme catalyzes the insertion of iron into the protoporphyrin IX (PfIX) ring. Accumulation of this molecule in erythrocytes, plasma, skin, and the liver is ultimately responsible for the clinical manifestations of EPP.
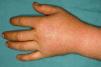
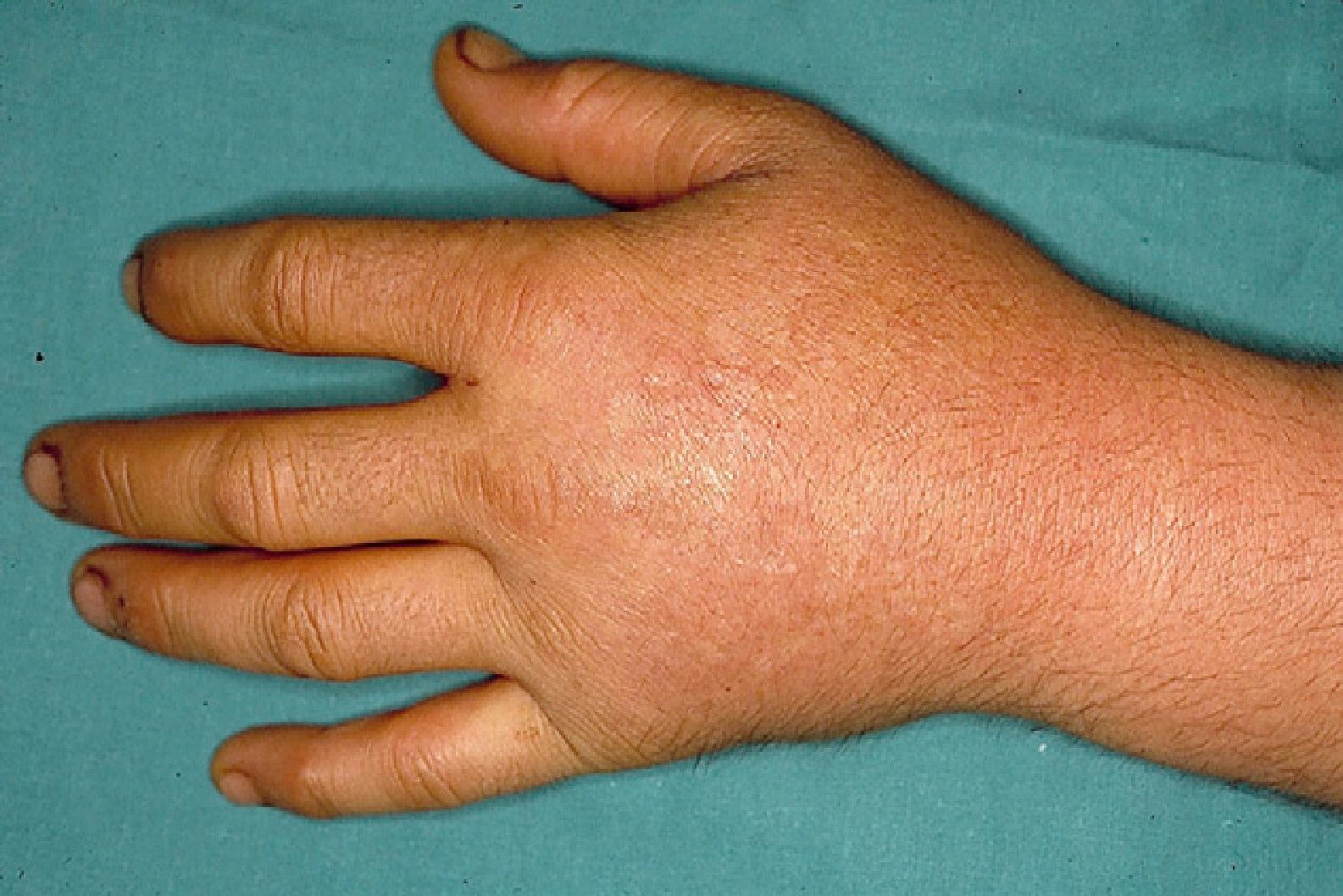
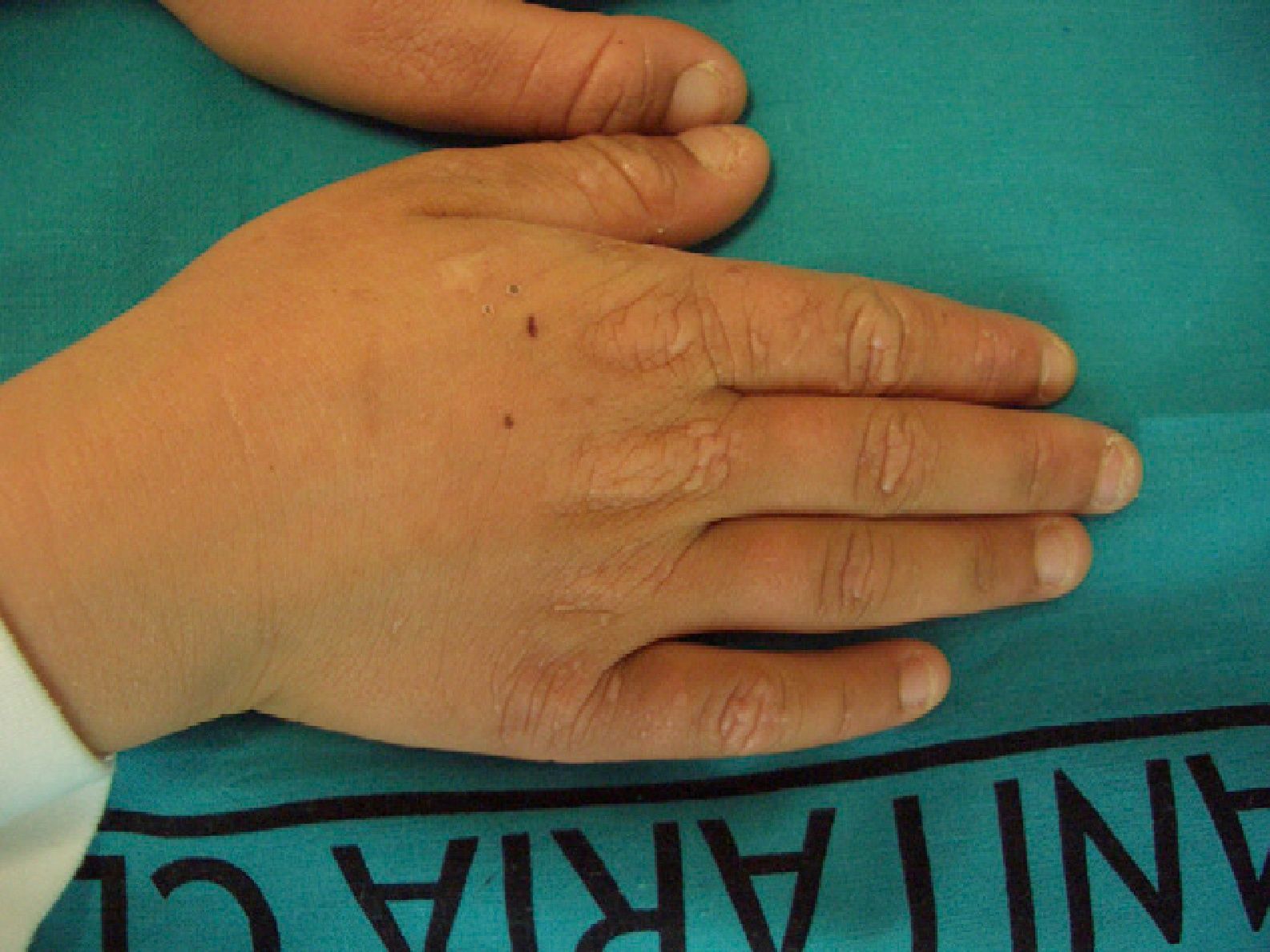
These manifestations are different to those of other cutaneous porphyrias. EPP presents as acute photosensitivity with erythema, edema, and a painful burning sensation, very similar to sunburn, although it only requires between 1 and 30minutes of exposure to sunlight (Fig. 2). Patients are therefore severely limited in their outdoor activities, with a marked negative impact on quality of life.1,4 Repeated exposure to sunlight can give rise to chronic lesions in sun-exposed areas. The lesions themselves, which appear as facial smallpox-like punctate scars and a waxy thickening of the skin, particularly in the perioral region and above the metacarpophalangeal joint, are not particularly disabling (Fig. 3).1,5
Given the intrinsic hydrophobia and liposolubility of PpIX, the molecule undergoes hepatic clearance in bile and feces. Excess PpIX in the bile canaliculi can cause cholestatic liver disease. Up to 25% of patients may experience cholelithiasis due to porphyrin gallstones, and up to 5% may develop severe liver disease leading to liver failure.6 In such cases, liver transplantation may be the only option.
Almost half the patients have anemia, which is normally mild and asymptomatic, with iron deficiency (microcytosis and hypochromia) refractory to iron supplementation.7
In biochemical terms, EPP is characterized by increased concentrations of free PpIX (not bound to zinc) in red blood cells and plasma. The plasma fluoresces with a peak emission around 634nm. Unlike the case of other cutaneous porphyrias, porphyrins are not detected in urine as PpIX is only eliminated in feces.8
In recent years, important molecular discoveries concerning EPP have been made, enabling the hereditary transmission mechanism in the autosomal forms to be elucidated. Subgroups of patients with genotypic and phenotypic characteristics have been identified and new forms of the disease caused by genes other than those that encode FECH have been found. The disease is therefore no longer considered monogenic but rather a heterogenous condition in which it is not always easy to establish a correlation between genotype and phenotype. Recent discoveries in the genetics of EPP have enabled a classification system based on molecular criteria to be designed, although this system has obvious clinical applications, particularly with regard to the development of severe liver disease (Fig. 4).
Autosomal Dominant Form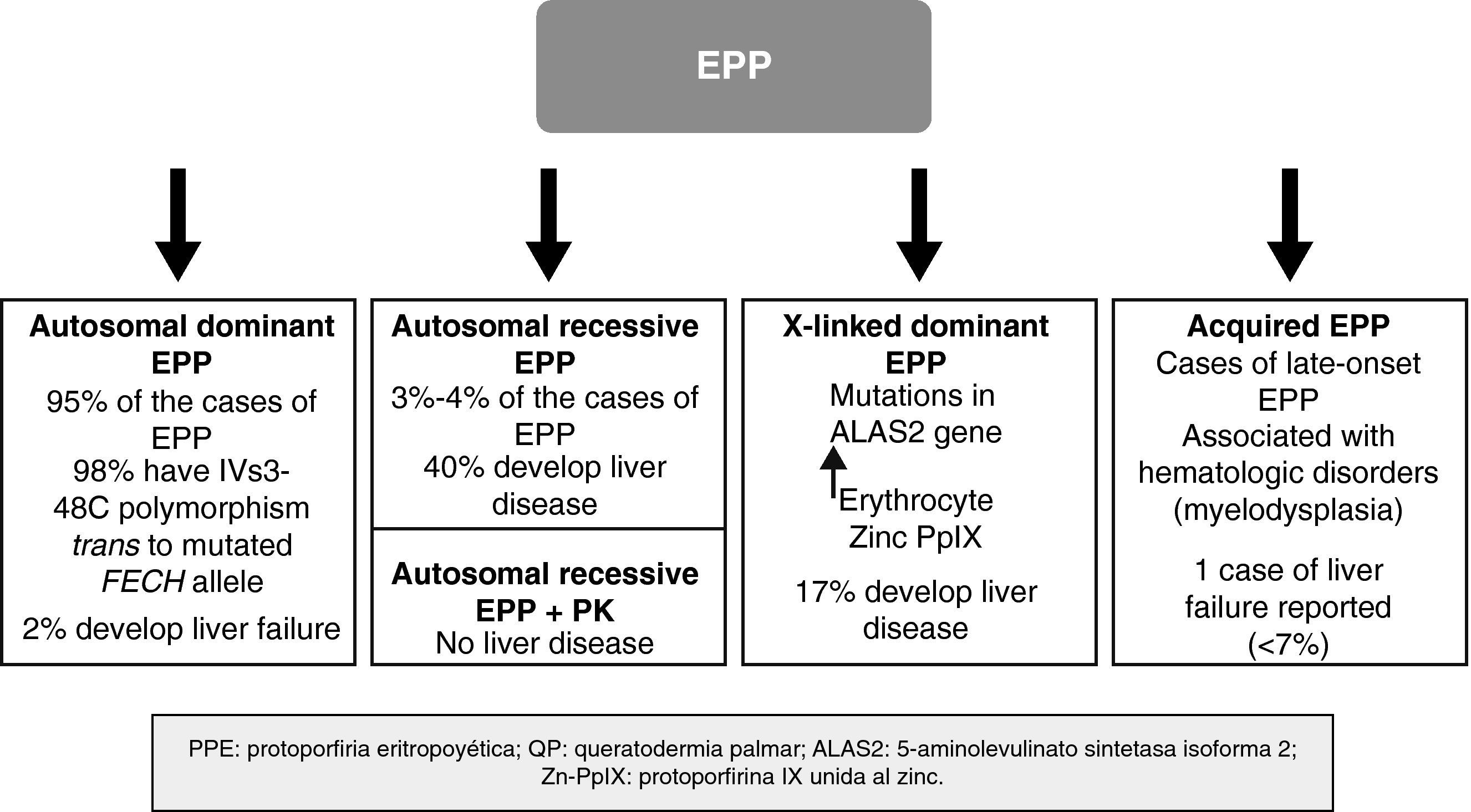
The autosomal dominant form is the most common form EPP and accounts for almost 95% of cases. It is caused by the transmission of a mutation in 1 of the alleles of the FECH gene, leading to a quantitative or qualitative change in the ferrochelatase enzyme. The degree of residual enzymatic activity varies among patients and family members with the mutation, leading to a pattern of autosomal dominant inheritance with incomplete penetration. Thus, residual enzymatic is around 50% of normal activity in carriers of 1 of the 2 mutated alleles but less than 35% in symptomatic individuals.9 For years, it was suspected that this additional decrease in enzymatic activity could be related to the presence of a common polymorphism in the nonmutated allele and that this would lead to expression of the gene below the critical threshold for manifestation of the disease.10,11 In 2002, Gouya et al.12 identified a polymorphism in intron 3 of the FECH gene, IVS3-48C, present in 98% of patients, as responsible for the low expression of the nonmutated FECH gene. The exchange of a single nucleotide, IVS3-48T>C, leads to a greater percentage of aberrant mRNA, which is rapidly degraded, in turn leading to decreased protein expression. This finding helped investigators understand the mechanism by which only some carriers of the mutated gene develop the disease. Thus, for EPP to develop, both the mutated allele and the low-expression allele (IVS3-48C) have to be transmitted. The prevalence of this allele varies greatly among different populations.13 Thus, for example, the prevalence of 43% found in Japan is much higher than that found in western Africa (<1%).14,15 By contrast, the prevalence of the low-expression allele appears fairly constant throughout Europe, with values of 13%, 11%, and 10.5% in the United Kingdom, France, and Spain, respectively.12,16,17
To date, it has not been possible to establish a relationship between specific mutations for the FECH gene and PpIX concentration, the age of onset of the disease, or the severity of the cutaneous manifestations. Nevertheless, there does seem to be a significant genotype-phenotype correlation between carriers of null-allele mutations (leading to complete loss of enzyme function) and severe liver disease.18,19
Autosomal Recessive FormApproximately 4% of patients with EPP are carriers of actual mutations in both alleles, and thus have the recessive form of the disease. Although only around 20 cases have been reported, this is the most common form of EPP in individuals who are not carriers of the low-expression allele.16
In clinical terms, these patients have the same dermatologic characteristics as the heterozygous patients. However, the risk of developing severe liver disease and liver failure requiring transplantation is much higher; close monitoring of liver function and protoporphyrin concentrations in plasma and feces is therefore recommended (Fig. 4).20
Recently, Holme et al.21 described a subgroup of 9 patients with recessive EPP (rEPP) with a characteristic palmar keratoderma. In all patients, keratoderma presented in early childhood. In 7 of these patients, onset of the lesion occurred before diagnosis of EPP. In all cases, the keratoderma had well-defined edges, without an erythematous border, and minimal spread. The severity of hyperkeratosis was highly variable but worsened on exposure to sunlight in all cases, and so showed a seasonal variation. In addition, all patients had thickening of the skin at the knuckles, a sign present in 35% of patients with EPP. In the biochemical analysis, a lower PpIX concentration in erythrocytes was observed in these patients compared to patients without keratoderma. Almost all patients were carriers of missense mutations (responsible for certain changes in amino acids that lead to the formation of an abnormal protein that nevertheless conserves a certain degree of functionality). No genotype-phenotype relationship could, however, be established as these mutations had also been reported in patients without keratoderma. Nevertheless, the most important finding was the absence of patients with liver involvement and palmar keratoderma, in contrast to the high percentage of patients (up to 42%) with severe liver involvement in the group with rEPP but no keratoderma.
Thus, the presence of palmar keratoderma at very young ages could be considered a clinical sign specific for rEPP, defining a subgroup at low risk of developing liver disease.
Dominant X-linked FormIn 2008, Whatley et al.22 described a new type of EPP in patients without mutations in the FECH gene. No FECH mutations were found in approximately 7% of the families with EPP studied at a molecular level. Within this subgroup, the investigators identified 8 families with members affected by a form of EPP clinically identical to that of patients with the dominant autosomal form (dEPP). However, differential characteristics were present: the PpIX concentration in erythrocytes was higher than in patients with dEPP; a much higher proportion of zinc PpIX was observed than in patients with the dEPP form; and a dominant hereditary pattern was linked to the X chromosome. This last finding prompted study of X-chromosome genes that encode important proteins in the heme biosynthetic pathway. This approach demonstrated that certain mutations in the aminolevulinic acid synthase 2 (ALAS2) gene were responsible for this new form of EPP.
ALAS2 is a specific erythroid isoform of 5-aminolevulinate synthetase (ALAS) and is essential for synthesis of hemoglobin in erythroid cells. Until recently, only mutations responsible for hereditary sideroblastic anemias had been reported. In the study of Whatley et al.,22 2 different deletions were detected (c.1706-1709 delAGTG and c.1699-1700 delAT) in exon 11 of the ALAS2 gene. These are responsible for alterations in amino acids 19 and 20 of the C-terminal extremity of the enzyme that causes structural change in the protein and in turn enhances its function. This is the first time a function-enhancing mutation has been identified in one of the enzymes involved in heme biosynthesis. With the increased functionality of the ALAS2 enzyme, which is the limiting enzyme of the metabolic pathway in normal conditions, ferrochelatase becomes the new limiting enzyme, leading to a greater accumulation of zinc protoporphyrin.
Of note is the high percentage of patients with X-linked EPP who have hepatic disease (17%). This suggests that X-linked EPP is associated with a greater risk of severe liver disease, by analogy with the autosomal recessive form.
Thus, abnormalities in the C-terminal region of ALAS2 lead to production of protoporphyrins in sufficient excess to cause photosensitivity and liver damage, even though ferrochelatase activity is completely normal.
Acquired Forms (Late Onset)Acquired EPP, with onset during adult life (age > 18 years) has been reported in relation with hematologic malignancies.23 Although it is a very uncommon entity, it should be considered in patients with adult-onset severe acute photosensitivity.
Fewer than 15 cases have been published and these were mainly associated with myelodysplastic syndromes, and the sideroblastic anemia subtype in particular.24 Patients with myelodysplasia show genetic instability that leads to the formation of complex chromosomal abnormalities in the myelodysplastic cell clone. It has been postulated that the development of EPP in these cases could be the result of clonal expansion of hematopoietic cells containing just 1 allele of the FECH gene, as a result of deletion of 1 of the 2 copies on chromosome 18.24,25
Although the prevalence of liver involvement associated with this form of EPP is not known, at least 1 case of fatal liver disease has been published.26
Treatment of Erythropoietic PorphyriaThe main therapeutic measure in EPP is still avoidance of sun exposure, even through window glass. Administration of high oral doses of beta-carotenes (30mg/kg/d) and the different types of phototherapy usually improve tolerance to light.27–29 More recently, melanocyte-stimulating hormone analogs, which also act as agonists of the melanocortin receptor, also appear to be useful as they enhance melanin synthesis, thereby increasing skin pigmentation and tolerance to natural light.30
The utility of oral administration of iron in an attempt to reduce the anemia present in many patients with EPP is still the subject of debate as cutaneous photosensitivity may worsen in some cases.31 However, in cases of severe anemia, endovenous administration of iron or even transfusion may be necessary.
Cholestyramine (4-16g/d) can be used to accelerate the elimination of protoporphyrins by the biliary route, with the corresponding reduction in both plasma levels and risk of liver involvement.32 Ursodeoxycholic acid and chenodeoxycholic acid have also been used for this purpose.33 However, the main complication of EPP is still cholestatic liver disease caused by protoporphyrin deposition, with the risk of acute irreversible liver failure requiring liver transplantation (Fig. 5). Since the first liver transplantation in a patient with EPP in 1979,34 more than 40 such transplantations have been reported, with an overall 1-year survival of 77% and a 5-year survival of 66%.35 We should remember, however, there are 3 major complications: a) phototoxic reactions induced by operating room lights, which may be avoided by covering the lights with yellow filters that block radiation below 460nm36; b) postoperative motor neuropathy with very high PpIX plasma levels37; and c) the recurrence of liver disease due to protoporphyria in the transplanted liver, which can be prevented by hematopoietic stem cell transplantation.35,38 Thus, in the event of severe and progressive liver disease, the possibility of a sequential liver and bone marrow transplantation should be considered or, ideally, bone marrow transplantation should be performed before liver failure occurs.35,39
Congenital Erythropoietic Porphyria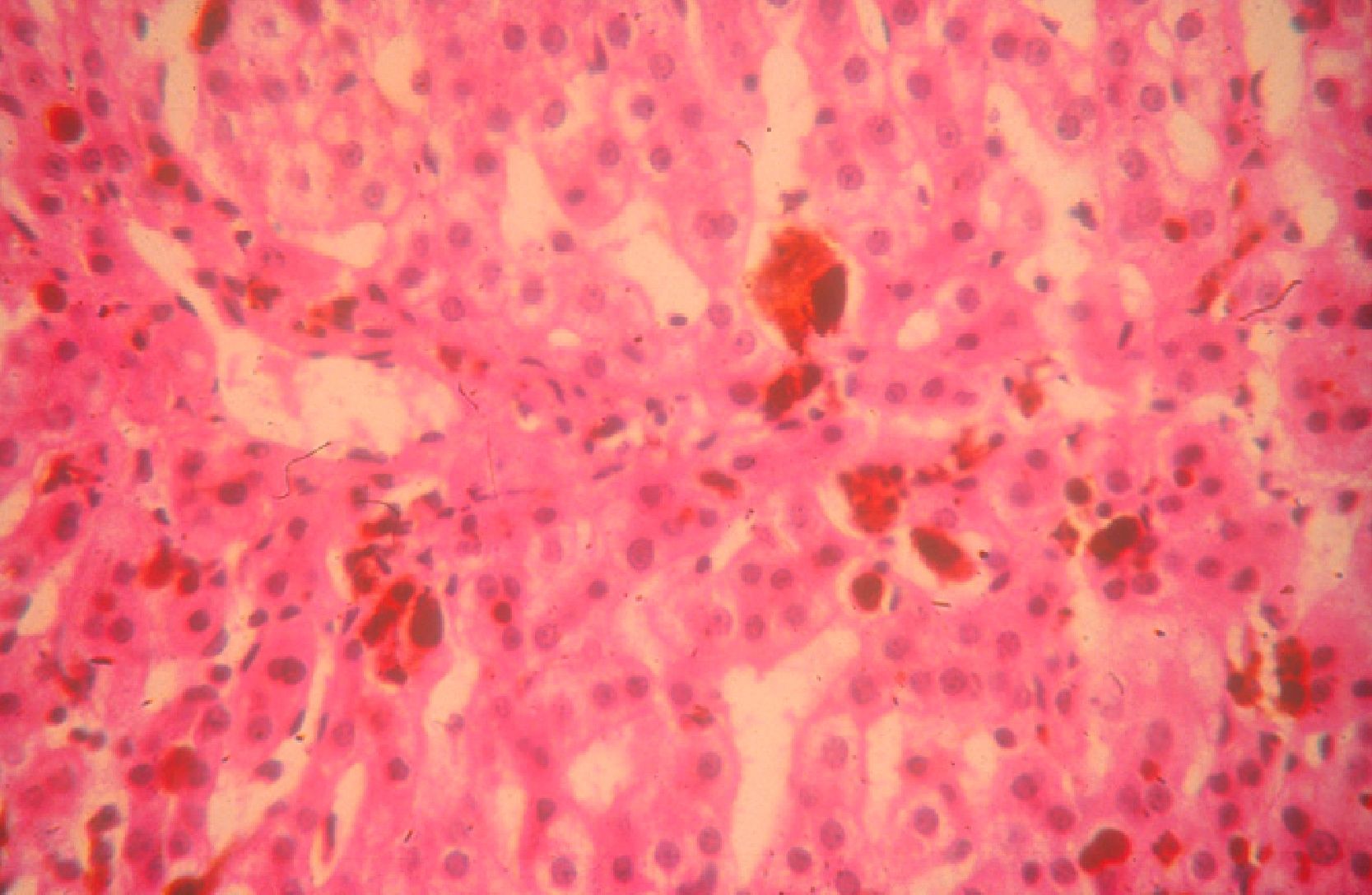
CEP or Günther disease follows a recessive autosomal transmission pattern. The disease is extremely rare, with approximately 150 cases reported in the literature.40 It is caused by deficient activity of uroporphyrinogen-III synthetase (UROS), which is encoded by the UROS gene on chromosome 10q25.3→q26.3.41
The clinical manifestations of CEP are heterogeneous and very variable, ranging from the onset of hydrops fetalis as a result of severe intrauterine hemolytic anemia to very mild forms in which there is hardly any skin involvement in adults (Fig. 6). Passing of dark (reddish) urine in a neonate may be the first key sign for diagnosis, although this sign is also seen in other porphyrias such as hepatoerythropoietic porphyria and recessive forms of acute intermittent porphyria.42
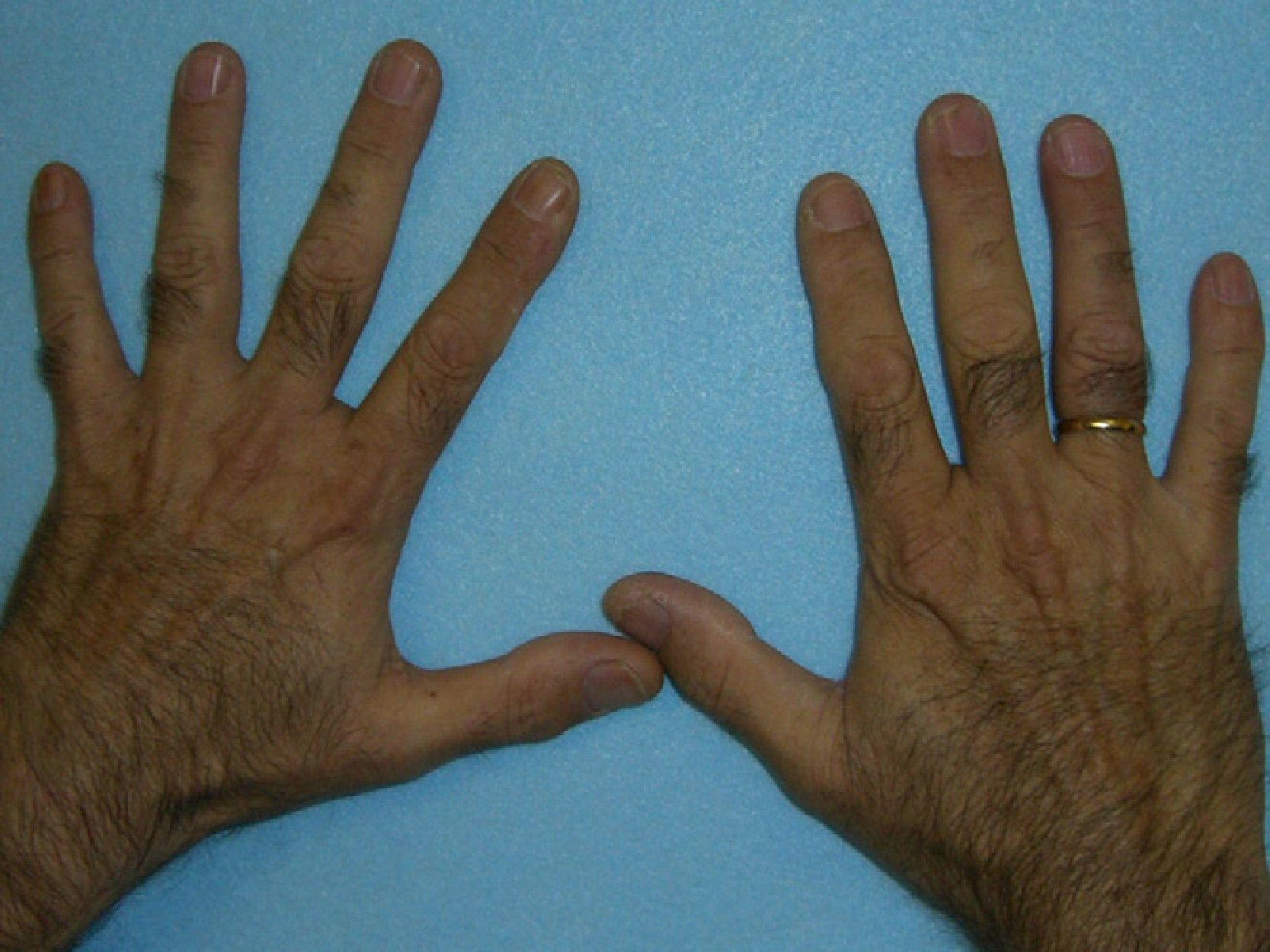
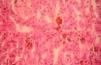
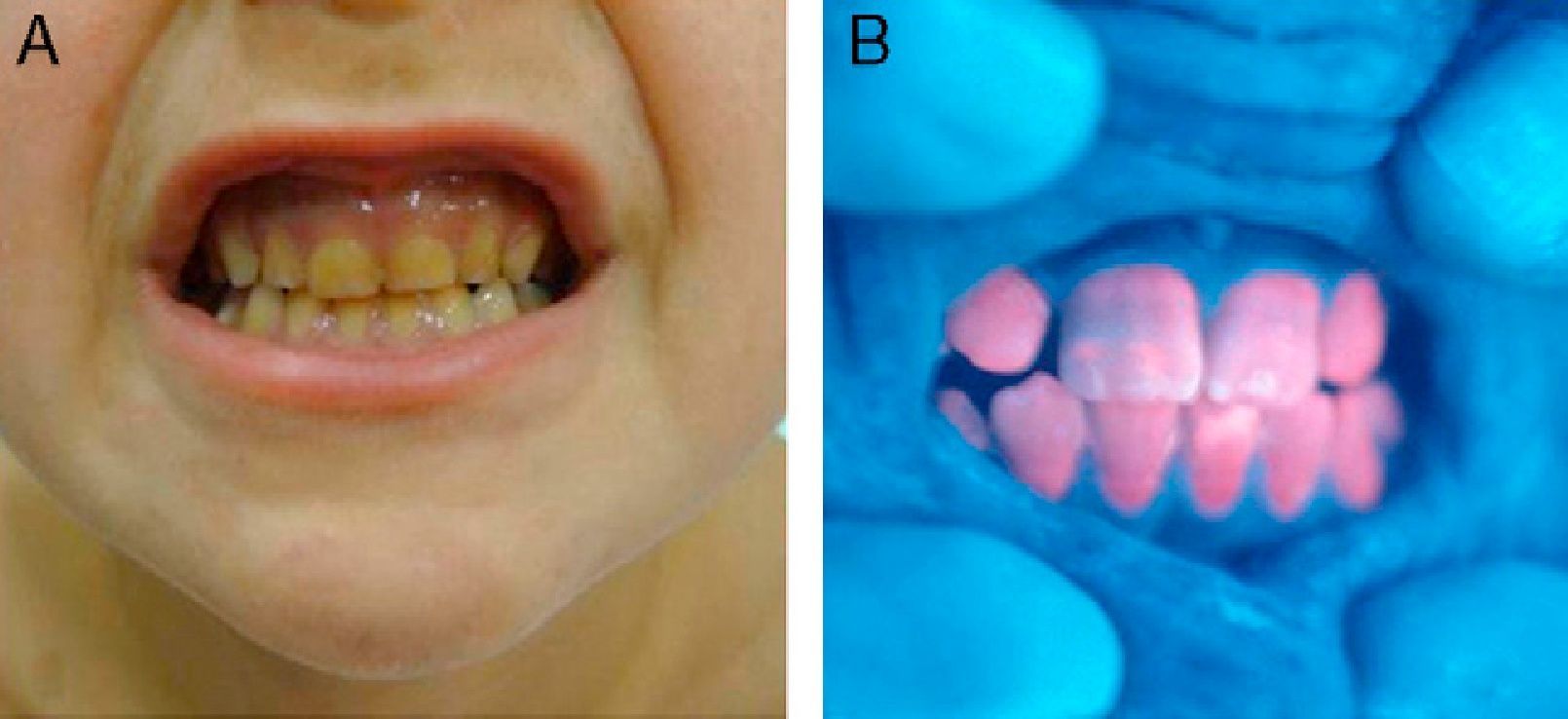
In general, patients with CEP have substantial photosensitivity already in early childhood. The recurrent appearance of blisters and secondary infection leads to scarring, mutilations, and deformities. Facial and generalized hypertrichosis is often severe. Porphyrin deposits in the cornea and developing teeth can lead to visual impairment and erythrodontia, respectively (Fig. 7).
Hemolytic anemia is common. Patients usually show anisocytosis, poikilocytosis, reticulocytosis, absence of haptoglobin, hyperbilirubinemia, and increased fecal urobilinogen. Secondary hypersplenism caused by inefficient erythropoiesis contributes to anemia and can also lead to leukopenia and thrombocytopenia. Anemia can be serious enough to require periodic transfusions.
CEP is the only porphyria in which type I porphyrins (uroporphyrin I and coporphyrin I) are produced in excess and can be detected in urine (uroporphyrin I) or feces (coporphyrin I). Red blood cells contain large amounts of uroporphyrin I and, to a lesser degree, coporphyrin I and zinc protoporphyrin (bound to zinc).
Forty-three different mutations of the UROS gene have been reported.43 The vast majority of these are missense mutations. The p.C73R mutation in exon 4 is the most common mutation, with an allele frequency of 30%; the homozygous form is associated with a severe phenotype. However, the majority of the remaining mutations have been found in single families or, at most, just a few families of CEP patients. In addition, although it is generally accepted that the degree of residual in vivo activity of the UROS enzyme determines the severity of the disease, there have been reports of patients with the same mutations in the UROS gene with very different phenotypes.44 As a result, it is very hard to establish a genotype-phenotype correlation in CEP. This strengthens the hypothesis that genes other than the UROS gene can act as gene modifiers in the disease. Two major recent discoveries illustrate this possibility. The first, published by Phillips et al.,45 was the description of the first case of CEP attributed to a mutation in the gene encoding the erythroid-specific transcription factor, GATA binding protein 1 (GATA-1), on the X chromosome (Xp11.23). This transcription factor is essential to ensure functions such as normal erythropoiesis, appropriate expression of globin genes, and appropriate development of megakaryocytes. But, in addition, it acts by regulating the expression of the UROS gene in developing erythrocytes. Thus, GATA-1 mutations can be responsible for CEP in the absence of UROS mutations and so GATA-1 probably acts as another modifier gene in some patients with UROS mutations. The second relevant discovery, reported by To-Figueras et al.,46 was that gain-of-function mutations in the ALAS2 gene, as described for X-linked EPP, may act as an aggravating factor resulting in more severe biochemical abnormalities and clinical manifestations in patients with UROS mutations. Thus, mutations in the ALAS2 gene corresponding to changes in the C-terminal region that increase enzymatic activity, along with mutations in the UROS gene that slow enzymatic kinetics at this level, lead to a greater accumulation of type I porphyrins and, as a result, more severe clinical manifestations.
Finally, we should remember that there are cases of acquired CEP, with onset during adulthood. Indeed, around 5 cases of CEP with onset after 18 years of age have been reported.47,48 These tend to show milder clinical manifestations and are often associated with thrombocytopenia and myelodysplasia, by analogy with acquired EPP.
Treatment of Congenital Erythropoietic PorphyriaThe 3 cornerstones of therapeutic management of patients with CEP are avoidance of photoexposure, meticulous care of cutaneous wounds, and blood transfusions and other hematologic support measures.
Other symptomatic treatments include hydroxyurea to reduce porphyrin synthesis in bone marrow,49 splenectomy to reduce the need for transfusions in patients with hypersplenism, and oral administration of activated charcoal to facilitate fecal excretion of porphyrins. However, many of these measures cannot be considered a long-term solution. Indeed, hematopoietic transplantation is the treatment of choice in patients with the most severe forms of the disease (transfusion-dependent patients).50 There have been approximately 15 reports of patients with CEP who have received allogeneic bone marrow transplantation, in most cases with clinical and biochemical cure of the disease.51 Although gene therapy is still not a real option for curing this porphyria, notable progress has been made in the experimental setting and so such an approach may prove useful in the medium term.52
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Darwich E, Herrero C. Novedades en las porfirias eritropoyéticas. Actas Dermosifiliogr. 2013;104:212–9.


