




HISTORIA CLÍNICA
Un varón de 75 años consultó por lesiones eritema-tosas violáceas asintomáticas de 7 meses de evolución, que se iniciaron en el escroto y se extendieron a muslos y piernas. Como antecedentes personales destacaba que era ex fumador y padecía un síndrome prostático. Las lesiones se iniciaron como máculas violáceas que pronto se transformaron en placas y nódulos.
EXPLORACIÓN FÍSICA
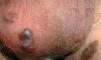
En el momento de la consulta presentaba máculas eritematosas violáceas, de 3 a 10 mm de diámetro, aisladas, localizadas en las piernas y muslos (fig. 1). Además tenía placas de color marrón-purpúrico y nódulos de color marrón-azulado en el escroto (fig. 2). No se observaron lesiones en otras localizaciones (cara, tronco o mucosas), el estado general era bueno y no se palpaban adenopatías ni visceromegalias.
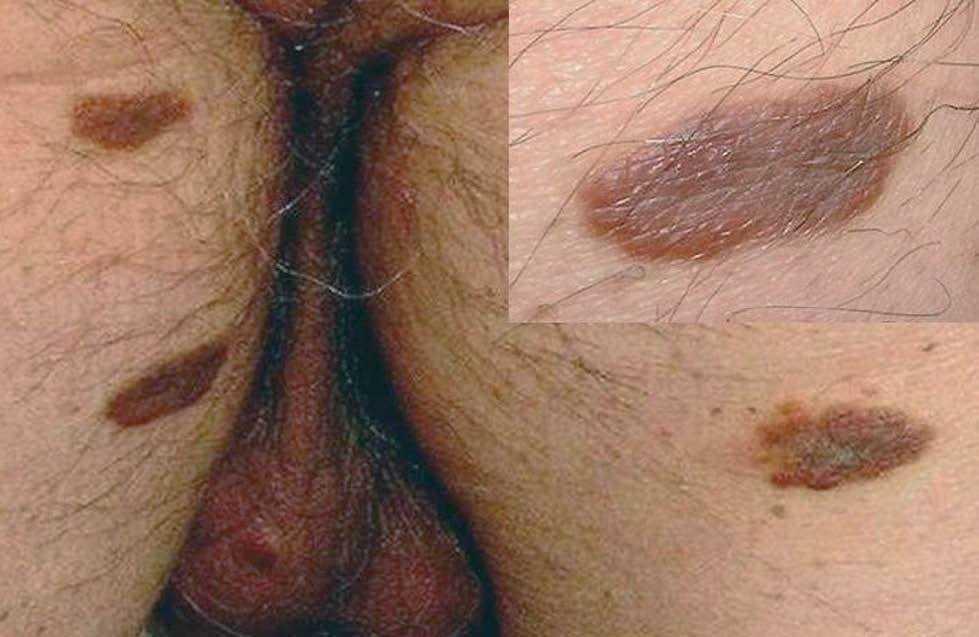
Fig. 1.—Máculas y pápulas marrones y eritematovioláceas localizadas en muslos.
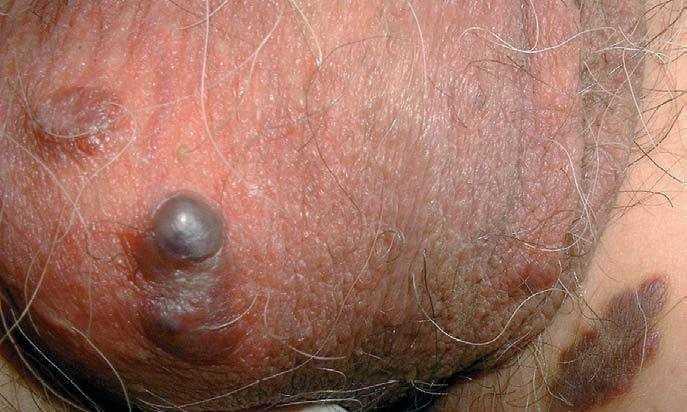
Fig. 2.—Nódulos violáceos en escroto.
EXPLORACIONES COMPLEMENTARIAS
El hemograma, estudio de coagulación, bioquímica básica, proteína C reactiva y velocidad de sedimentación fueron normales. Las serologías frente al virus de la inmunodeficiencia humana (VIH) y a los virus de la hepatitis A, B y C fueron negativas. El recuento de sub-poblaciones linfocitarias fue normal, así como el estudio cardiológico (electrocardiograma, ecocardiografía y ventriculografía isotópica de equilibrio), la radiografía de tórax, la ecografía abdominal y la tomografía computarizada toracoabdominal. Se realizó una biopsia «en sacabocados» de 5 mm de una placa del muslo izquierdo (figs. 3 y 4).
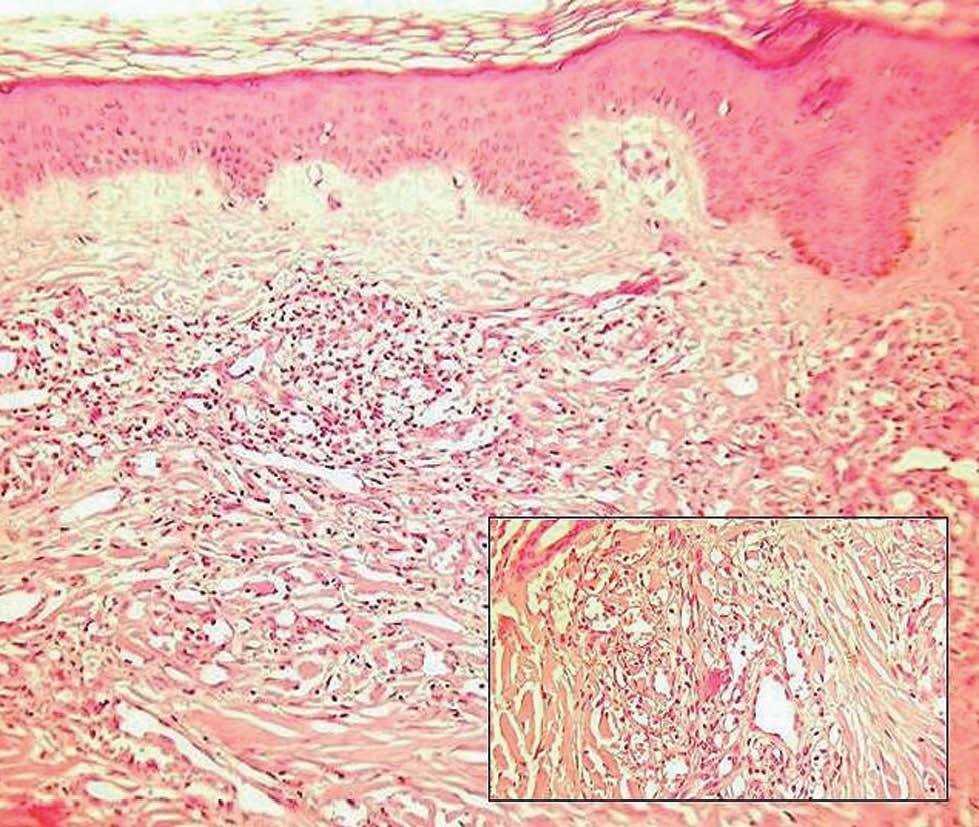
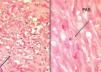
Fig. 3.—Epidermis ortoqueratósica y dermis con neoformación vascular e infiltrado inflamatorio. (Hematoxilina-eosina, ×10.) En el recuadro se observa la presencia de abundantes vasos de diferente tamaño con células atípicas, más abundantes en la parte superior izquierda. (Hematoxilina-eosina, ×40.) Obsérvese la presencia de pigmento hemosiderínico junto a la neoformación vascular.
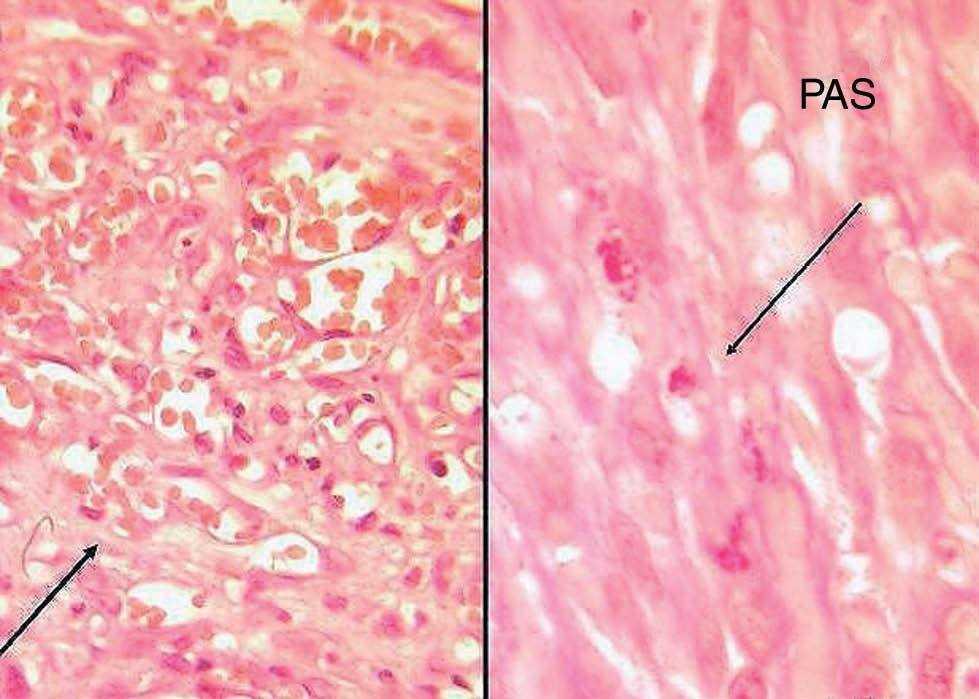
Fig. 4.—A) Capilares con células endoteliales atípicas. (Hematoxili-na-eosina, ×40.) B) Cuerpos de inclusión PAS+. (PAS, ×40.)
DIAGNÓSTICO
Sarcoma de Kaposi clásico.
HISTOPATOLOGÍA
Encontramos una epidermis ortoqueratósica y en la dermis se observaba una neoformación vascular con vasos de distinto tamaño y células atípicas en su interior. Llamaba la atención la presencia de un infiltrado inflamatorio abundante. Próximo a los vasos neoformados se apreciaba la presencia de pigmento hemosiderínico. Con la tinción de ácido peryódico de Schiff (PAS) se tiñeron algunos cuerpos de inclusión (figs. 3 y 4).
EVOLUCIÓN Y TRATAMIENTO
Con el diagnóstico de sarcoma de Kaposi clásico limitado a piel se instauró tratamiento con adriamicina liposomal en dosis de 85 mg cada 4 semanas. Después de cinco sesiones de quimioterapia dejaron de aparecer nuevas lesiones y las previas se aplanaron, disminuyendo de tamaño y adquirieron tonalidades más pálidas, que incluso llegaron a desaparecer por completo. En los controles periódicos realizados durante un año y medio el paciente ha permanecido asintomático.
COMENTARIO
El dermatólogo húngaro Moritz Kaposi, entre otras muchas enfermedades describió en el año 1872 los primeros 5 casos de sarcoma idiopático múltiple pigmentado, conocido en la actualidad como sarcoma de Kaposi1 .
Se definen cuatro variantes en función de las características clínicas y el curso de la enfermedad: el clásico, mediterráneo o crónico, que corresponde a nuestro caso; el endémico, linfangiopático o africano; el asociado a trasplantes o iatrogénico y el epidémico, descrito en la década de los años 1980 en relación al sida.
El clásico inicialmente afecta a personas de origen judío o mediterráneo. Representa entre el 0,02 a un 0,05 % de los tumores malignos. Aparece con más frecuencia en varones (relación hombre:mujer 10-15:1) de 50 a 70 años. Se presenta como máculas, placas y/o tumores rojos, violáceos o marronáceos que, en general, sólo afectan a las extremidades inferiores, sobre todo los tobillos, los talones y las plantas de los pies. Otras localizaciones son infrecuentes2 . El linfedema y la insuficiencia venosa de la extremidad afectada son complicaciones comunes. En general, el curso de la enfermedad es benigno e indolente, y durante años se observa un crecimiento lento de las lesiones y aparición gradual de nuevos elementos. En casos de larga evolución pueden afectarse el tracto gastrointestinal y los ganglios linfáticos3 . En un tercio de los casos se asocia a neoplasias sistémicas, la mayoría de las veces a linfoma no hodgkiniano4,5 , en relación con el grado de inmunodepresión subyacente.
La etiopatogenia del sarcoma de Kaposi siempre ha sido un enigma6 . La asociación con factores genéticos aún no ha sido confirmada. Su relación con determinados marcadores HLA sólo se ha observado en algunos subtipos de sarcoma. En el año 1966, Williams y Williams implicaron por primera vez a un agente infeccioso en la etiología de la enfermedad, que años más tarde Giraldo et al, identificaron como un tipo de herpes vi-rus3,4 . La primera relación entre sarcoma de Kaposi y el virus denominado herpes virus humano tipo 8 (HVH8) o virus asociado a sarcoma de Kaposi (KS-HVH) fue establecida por Chang et al. en 19947,8 . Se demuestra su participación en el 95-100 % de sarcoma de Kaposi, con independencia del subtipo de la enfermedad, de la raza y de la localización geográfica. Desde entonces, el HVH8 se ha implicado en otras enfermedades, como la de Castleman, el linfoma primario de cavidades y el síndrome POEMS. Aunque el significado etiopatogénico de estas asociaciones es incierto, los conocimientos actuales refuerzan el concepto de sarcoma de Kaposi como una enfermedad sistémica en la que hay una proliferación celular inducida por el propio virus9 . Sin embargo, la secuencia genética del virus ha revelado la presencia de oncogenes, lo que podría explicar su potencial para inducir una trasformación maligna9 .
Las características histológicas son muy parecidas en todos los tipos de sarcoma de Kaposi, pero dependen del estadio clínico. En la fase macular predomina el componente vascular; en la dermis se observan espacios vasculares irregulares que disecan focalmente las estructuras vasculares preexistentes (signo del promontorio) y los haces de colágeno. Las paredes de dichos espacios son finas y están delimitadas por células endoteliales prominentes. Es habitual que existan infiltrados linfoplasmocitarios perivasculares, hematíes extravasados y depósitos de hemosiderina. En la histología de las pápulas, placas y nódulos predomina el componente celular, con haces entrelazados de células fusiformes y hendiduras vasculares poco definidas. Las células fusiformes pueden mostrar grados diferentes de pleomorfismo y, en general, escasas figuras mitóticas. Expresan CD34 y, de forma inconstante, el CD31 y el antígeno relacionado con el factor VIII. Es común la presencia de glóbulos eosinofílicos intra y extracelulares que se tiñen con la tinción de PAS. La naturaleza exacta del sarcoma de Kaposi es, desde su descripción, motivo de controversia, ya que para algunos autores se trataría simplemente de una hiperplasia, mientras que para otros sería un verdadero proceso neoplásico10 .
El diagnóstico diferencial hay que establecerlo, según el aspecto clínico, con tumores pigmentados, vasculares, dermatofibroma, granuloma piógeno, tumor glómico, angiomatosis bacilar y linfomas cutáneos, entre otros. Las enfermedades angioproliferativas clasificadas como seudosarcoma de Kaposi11 tienen una histología difícil de distinguir del verdadero sarcoma.
No existe un tratamiento tipificado, sino que depende de la extensión de la enfermedad y del estado general del paciente. Clásicamente se ha empleado la radioterapia, tanto en lesiones aisladas como en la enfermedad diseminada con buenos resultados y mínimos efectos secundarios. La criocirugía y la exéresis quirúrgica son buenas alternativas si las lesiones están muy localizadas. La quimioterapia también es eficaz, se dispone de una gran variedad de agentes y pautas, aunque no existe ningún estudio prospectivo que demuestre la eficacia de unos esquemas terapéuticos respecto a otros. La norma actual es el empleo de antraciclina liposómica, como tratamos a nuestro paciente, ya que esta presentación galénica prolonga la vida media del fármaco, aumenta su concentración en el tumor, y disminuye su toxicidad. Otras alternativas, más estudiadas en pacientes con sida, son el ácido 9-cis retinoico tópico12,13 y los interferones, con una tasa de respuesta objetiva del 40 % en pacientes con sarcoma de Kaposi epidémico. No se dispone de datos al respecto en el caso del tipo clásico, posiblemente por la baja prevalencia de la enfermedad, la benignidad del cuadro y la respuesta buena a otros tratamientos convencionales4 .
Aunque lo habitual es que el sarcoma de Kaposi clásico siga un curso benigno, es importante realizar controles periódicos para diagnosticar precozmente una progresión rápida de la enfermedad y la posible afectación extracutánea.
Correspondencia:
Cristina Serrano. Servicio de Dermatología. Hospital Clínico Universitario San Cecilio. Avda. Madrid, 9. 18012 Granada. España. crisefa@yahoo.es
Recibido el 2 de julio de 2004. Aceptado el 28 de noviembre de 2004.


